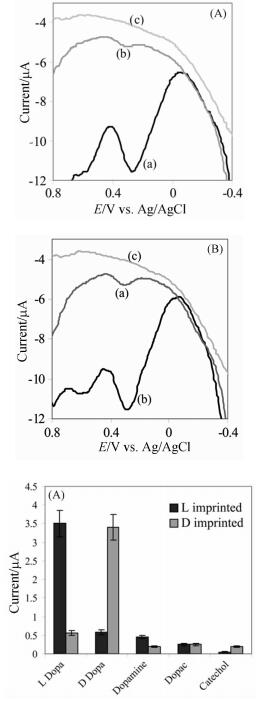
 图 1
dopa印迹膜的SWV曲线[39]
Figure 1.
SWV of imprinted films [39]
图 1
dopa印迹膜的SWV曲线[39]
Figure 1.
SWV of imprinted films [39]
引用本文:
张亚会, 刘刚, 徐慧. 电化学方法手性识别的研究进展[J]. 化学通报,
2017, 80(8): 699-707.

Citation: Zhang Yahui, Liu Gang, Xu Hui. Progress in Electrochemical Methods for Chiral Recognition[J]. Chemistry, 2017, 80(8): 699-707.
Citation: Zhang Yahui, Liu Gang, Xu Hui. Progress in Electrochemical Methods for Chiral Recognition[J]. Chemistry, 2017, 80(8): 699-707.
电化学方法手性识别的研究进展
English
Progress in Electrochemical Methods for Chiral Recognition
Abstract:
At present, chromatographic, spectroscopic, microscopic and electrochemical methods have been successfully used in the qualitative and quantitative study of separation and analysis of chiral compounds, and many significant progresses have been made over the past years. Among these approaches, electrochemical methods have attracted a lot of attention owing to the advantages of high sensitivity, simple instrument and fast detection. In this article, the research progress of electrochemical method for chiral recognition was reviewed, and the prospects of this field were also put forward.
-
Key words:
- Electrochemical sensor
- / Chiral recognition
-
自然界中有许多分子具有互成镜像但不能重叠的两种结构形式,如同人的左右手一样,它们之间彼此不能重合的性质称为手性。这两种结构的手性分子可以互称为手性异构体,也可称为对映体或光学异构体[1],它们在生物体内的药理活性、代谢过程、代谢速率及毒性等存在显著差异[2, 3]。在实际应用中,往往一种对映体对人体起到药理的作用而另一种对映体则不起作用或有毒副作用[4],例如19世纪60年代初期的“反应停事件”[5]。手性识别是一种手性分子区分另一种手性分子对映体的能力,就像手跟手套的关系。手性化合物的识别研究在医药[6~8]、农药[9]、食品添加剂[10]等许多方面具有重要的理论意义和应用前景[11]。因此,发展简单、快速、准确的手性识别方法成为近几年来分析化学的热点和前沿方向[12]。目前,许多方法已成功用于手性识别并取得了一些进步,其中电分析方法因具有灵敏度高、仪器简单、检测快速等特点而得到迅速发展。
1 手性识别方法
手性化合物的拆分和识别是手性分析的重要内容,色谱法、光谱法、显微技术和电化学方法等已用于手性化合物的定性和定量研究,并取得了一定的研究进展。
1.1 色谱法
目前常用于手性化合物分离的色谱方法主要包括高效液相色谱(HPLC)[13~18]、毛细管电泳(CE)[19~22]、气相色谱(GC)、薄层色谱(TLC)和超临界液体色谱(SFC)等。色谱分离分析方法一般采用直接法和间接法两种形式。直接分离是通过手性固定相或者手性添加剂的方法直接分离手性对映体;间接法是通过手性衍生化试剂与被分离物质反应,利用对映体的两种构型与手性衍生化试剂形成的复合物的物化性质不同来实现其在非手性固定相上分离[23]。
1.2 光谱法
用于手性识别的光谱方法主要有核磁共振(NMR)、紫外-可见光谱(UV-Vis)、红外光谱(IR)、荧光光谱、质谱(MS)和圆二色谱(CD)等[24~28]。这些方法是基于手性选择剂与手性化合物异构体之间形成不同的非对映异构体,或手性选择剂对手性异构体吸附效果的差异而导致光谱信号发生变化,从而达到手性识别的目的。
1.3 显微技术
扫描电镜(SEM)、原子力显微镜(AFM)和扫描隧道显微镜(STM)等技术[29~31]已用于手性识别。这类方法不仅在分子水平上直接对表面手性现象和手性结构进行研究,而且可以深入了解分子吸附、分子间相互作用、手性分离与识别等科学和实际应用的问题。
1.4 电化学法
电化学传感器具有仪器微型化、操作方便快速、在线、实时、低成本、灵敏度高、环保等优点。将手性识别与电化学方法结合形成的手性电化学传感器通过不同的电化学技术(例如循环伏安法(CV)、差分脉冲伏安法(DPV)、电化学交流阻抗技术(EIS)等)研究手性选择剂与目标手性分子间相互作用后电化学信号的差异[32]。常见的手性选择剂有多糖(壳聚糖)、蛋白质(牛血清蛋白(BSA)、γ-球蛋白、胶原蛋白等)、大环状和笼状分子(环糊精)及人工合成的分子印迹聚合物等。电化学方法检测手性化合物时常结合SEM、透射电镜(TEM)、UV-Vis、热重分析(TGA)、FT-IR等技术表征传感器构建的过程。这些技术辅助电化学方法用来表征修饰电极材料的结构和形貌。
目前,HPLC可以有效分离和识别手性化合物,但缺点是每次分离的产品量很少,成本较高,效率低。CE虽操作简单,但只可用于分离低浓度的对映异构体,其他的如GC等技术操作较简单,但使用条件苛刻,限制条件较多,仪器比较昂贵。电化学手性传感器的设备成本较低,而且在设计、制造、维护方面要比HPLC简单。但因传感器检测只含有一个吸附解吸过程,这就要求手性识别过程必须十分有效地进行,传感器的寿命提高也是关键,这对电化学传感器的设计是很大的挑战。
2 基于分子印迹技术的手性电化学传感器
印迹技术(MIT)是一种分子识别技术,其理论源于Fischer的酶和底物作用的“锁钥模型”、Pauling的抗体形成学说和Dickey的“专一性吸附原理”。近年来,人们已经开始利用MIT专一性识别的特点来手性识别一些小分子,如氨基酸[33]、单糖[34]等,这无疑为手性识别开辟了一条崭新的道路。MIT的基本原理是在印迹分子(模板分子)存在下,将带有特殊官能团的单体(功能单体)与过量的基质单体进行聚合反应,因模板分子的存在,聚合过程中单体本身所带的官能团会由于与模板分子相互作用(共价键作用或非共价键作用)的需要进行调整,并形成特定的空间构象和高度交联的聚合物即分子印迹聚合物(MIP)。聚合完成后通过洗脱除去模板分子,聚合物主体上就形成了与模板分子空间匹配的具有多重作用位点的空穴,其大孔结构使模板分子可以在聚合物中进行分散。这种MIP对印迹分子及与其结构相似的客体分子具有较高的特异性结合能力,结合的强弱依赖于MIP和客体分子大小及形状的匹配度[35]。
早在20世纪90年代就有将MIP的“记忆”效应用于分子识别的报道。2003年之后,基于MIP电化学传感器的分离及分子识别技术得到广泛研究。MIP的突出特点是对被分离物或分析物具有高度的选择性,同时其具有良好的物理化学稳定性,耐高温、高压、酸碱、有机溶剂等恶劣环境的影响,容易保存、制备简单并可大量生产[36]。
1980年,Damen等[37]利用共价键作用合成了对手性分子具有识别能力的聚合物材料。他们将叔丁氧羰基-L-脯氨酸(Boc-L-proline)模板分子与乙烯基氯甲苯作用生成酯类,加入交联剂聚合,用三氟乙酸和溴化氢混合物洗脱模板分子,最后与L、D型的混合物作用。再次水解后,溶液中L-构型的脯氨酸衍生物要多于D构型,说明该聚合物对Boc-L-proline具有一定的选择吸附能力。
1984年,Andersson等[38]以乙烯基苯甲酸(PVB)为功能单体、二乙烯苯(DVB)作交联剂、L-苯丙酸乙酯(L-pheOET)为模板分子,合成非共价型MIP。PVB中的—COOH与L-pheOET中的—NH2之间存在的离子作用使得该聚合物对L-pheOET有一定的选择性,这是最早将MIP应用于氨基酸衍生物手性分离的研究报道。
2005年,Fireman-Shoresh等[39]将四甲氧基硅烷(TMOS)和苯基三甲氧基硅烷(PTMOS)功能单体与乙氧基乙醇、盐酸和水混合反应,再加入模板分子L-或D-多巴(L-dopa、D-dopa)、R-或S-N, N’-二甲基二茂铁苯胺(R-Fc、S-Fc)反应得溶胶溶液。通过旋涂法在电极表面制得手性分子印迹溶胶-凝胶膜(厚度约70nm)。CV法和方波伏安法(SWV)证明两类印迹膜对dopa和Fc对映异构体都有良好的手性识别能力,受结构相似化合物的干扰都可忽略(如图 1)。与其他邻苯二酚衍生物相比,dopa印迹膜灵敏度更高,选择性更强,可检测1nmol/L的dopa;Fc印迹膜能检测含量为2×10-6的目标分子。
2007年,Sekine等[40]研究不同有机溶剂(甲苯、环己烷、吡啶、二甲基甲酰胺)对MIP手性识别过程中性能的影响。先合成苯丙氨酸苯胺(PAA),然后以甲基丙烯酸(MAA)为功能单体、乙二醇二甲基丙烯酸酯(EGDMA)为交联剂、偶氮二异丁腈(AIBN)为引发剂、L型或D型PAA分别作为模板分子,采用原位引发聚合法在ITO电极表面成功接枝上MIP膜,洗脱模板分子,得到L-PAA或D-PAA印迹聚合物膜修饰的电极(LIP-ITO和DIP-ITO)。CV研究结果表明,在非极性溶剂电解液中,由非极性有机溶剂中合成的聚合物膜修饰电极对分析物测量时电流响应信号最强。
2004年,Liao等[41]利用CV电聚合得到L-组氨酸(L-His)印迹的聚丙烯酰胺膜,并采用EIS和压电石英晶体技术对其进行表征。所构建的MIP仿生传感器可用于L-His的手性识别和His对映异构体的手性分离。Prasad等[42]制备的两性离子/L-His印迹的MIP可选择性识别微量L-His,检测限为0.128ng/mL。该方法简单、快速,色氨酸(Trp)、酪氨酸(Tyr)和谷氨酸(Glu)等对检测的干扰可忽略,但该方法存在一定环境风险。
2011年Prasad等[43]将Cu2+-L-His或Cu2+-D-His配合物为模板分子的复杂MIP(CIP)用于选择性识别痕量L-His。先后将溶胶-凝胶液(包含三甲硅烷基环戊二烯、乙醇、盐酸、多壁碳纳米管(MWCNTs))和N, N二乙基氨基二硫代甲酸钠修饰于石墨电极(PGE)表面,获得光敏感膜修饰的电极;光照后,将L-His、硫酸铜、磷酸单(2-丙烯酰氨基-乙基)酯、EGDMA和MWCNTs以及二甲基亚砜(DMSO)混匀,旋涂法修饰在电极表面,紫外光照,洗脱His后获得CIP修饰电极(CIP/PGE)。通过CV法研究了Cu2+对模板聚合物选择性结合L-His的调节作用,发现Cu2+的存在增强了L-His氧化峰的响应信号,赋予L-His与单体之间强的相互作用及电活性。
2013年,Prasad等[44]将金属离子配合物MIP电化学传感器用于手性识别焦谷氨酸(PGA)对映异构体。首先,将L-PGA或D-PGA模板分子、5-甲基-2-噻吩羧酸(5-MTCA)单体和CuSO4溶液(摩尔比1:2:1) 反应得Cu2+-模板分子-单体的配合物预聚溶液,然后通过CV电聚合法在PGE表面得MIP膜。采用示差脉冲阳极溶出伏安法(DPASV)研究了L-PGA和D-PGA在不同修饰电极上的识别效果。Cu2+对PGA的电催化作用使该金属配位分子印迹聚合物对模板分子具有高亲和性和选择性,使键合位点与模板分子更匹配。水相中检测PGA同分异构体的线性范围为2.8~170.0 ng·mL-1,检测限为0.77ng·mL-1(S/N=3)。对实际样品(尿、脑髓液、血浆)中PGA异构体进行检测时灵敏度高且干扰小,线性范围为1.3~180.0 ng·mL-1。该方法可检测水相和实际样品中痕量的L-和D-PGA,在临床医学方面有重大意义。
2014年,Sajini等[45]以乙烯基功能化的羧酸化多壁碳纳米管(Fun-f-MWCNTs)为基底,在其表面以D-扁桃酸(D-MA)为模板分子、4-乙烯基吡啶为功能单体、EGDMA为交联剂热聚合一层MIP膜,可用于手性识别D-MA。该f-MWCNTs-MIP修饰电极检测D-MA时具有良好的选择性和稳定性,与本体聚合反应获得MIPs-f-MWCNTs和NIPs-f-MWCNTs(无印迹分子存在)相比,对D-MA分子有更好的吸附性能。
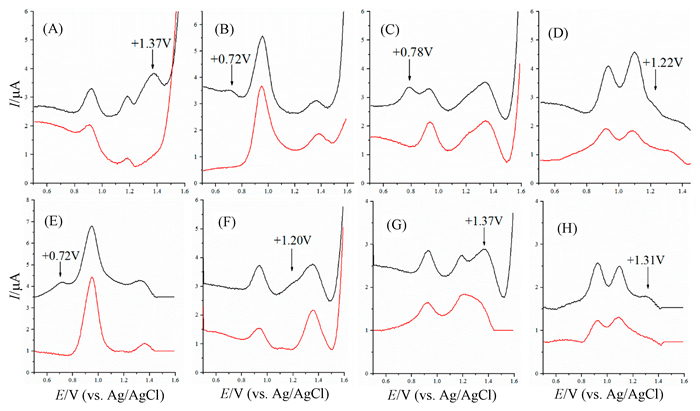
2015年,Iacob等[46]将MIP电化学传感器用于几种β-受体阻滞剂对映体的手性识别。以R(+)阿替洛尔(ATNL)或R(+)心得安(PRNL)作为模板分子、季戊四醇作交联剂、MAA为功能单体、乙腈为致孔剂、4-氰戊酸为引发剂,在四丁铵六氟磷酸电解液中,采用CV电聚合法在干净的碳糊电极(CPE)表面修饰MIP膜,通过改变电势扫描周期数来控制聚合物膜的厚度为小于10nm,得到MIP/CPE。根据DPV图中对映体峰电流值及氧化峰的位置,来区别四种β-受体阻滞剂及它们的对映异构体。结果表明,对于四种β-受体阻滞剂,R型异构体电流信号要强于S型(如图 2)。对ATNL的对映异构体的检测范围为1.88×10-7~1.88×10-5 mol/L。这是首例电化学手性传感器用于特异性识别β-受体阻滞剂对映体的报道。对在制药和生物医学领域寻求简单、经济和快速的旋光异构体定量分析方法有重要启示意义。
分子印迹导电聚合物(MICPs)由于本身具有电催化的特点,能增强检测信号,响应更迅速,性能更稳定;而且导电聚合物都具有高共轭性,因而具有可逆的化学、电化学和物理性能,并且这些特性能通过掺杂/脱掺杂的过程表现出来。在合成过程中避免了有机溶剂和复杂材料的大量使用,污染较低[47]。
2000年,Deore等[48]将由电位诱导法制备的分子印迹导电聚吡咯(PPy)膜用于分离Glu光学异构体,选择性分离系数可高达10。其缺陷在于洗脱模板分子的过程需使用电位调控法来改变PPy骨架上的电荷,且在吸附Glu光学异构体时使用的是溶剂富集法,此法只能用于少量光学异构体的分离。但该研究对MICPs分离和识别手性物质奠定了良好的基础。
2008年,Syritski等[49]通过电聚合法制备分子印迹PPy膜用于手性识别L-天冬氨酸(L-Asp)。他们将晶体电极浸入到吡咯(Py)为单体、L-Asp为模板分子的三种不同支持电解质水溶液(pH为6的弱酸、含1mol/L NaOH的pH为11的强碱、含聚对苯乙烯磺酸钠(NaPSS)的pH为6的弱酸溶液)中,恒电流聚合法得三种不同的L-Asp印迹PPy膜。采用电化学石英晶体微天平(EQCM),通过电势和质量的变化,研究这三种L-Asp印迹PPy膜的性能及其过氧化后的性能。SEM结果显示过氧化改变了PPy膜的形貌。在pH 6的溶液中电沉积于电极上的膜没有对映体选择性,而在含pH 11的强碱溶液中能形成平滑均一的L-Asp/PPy膜。而且,其在过氧化/去掺杂之后表现出相当好的对映体选择性,且测量的灵敏度受到膜的掺杂水平的影响。
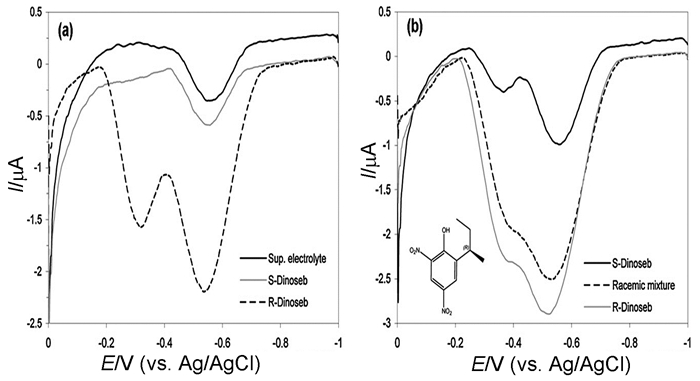
2011年,Basozabal等[50]用樟脑磺酸(CSA)诱导的聚苯胺(PANI)手性伏安传感器来手性识别地乐酚(dinoseb)对映异构体。以R-CSA或S-CSA和苯胺的混合溶液为聚合反应介质,CV电聚合法获得R-CSA-PANI和S-CSA-PANI修饰电极。分别用于特异性识别外消旋混合物中dinoseb对映异构体(图 3),具有良好的灵敏度和重现性(RSD < 10%和20%),选择性强。该方法可以扩展到外消旋混合物中其他对映异构体的识别。
2013年,Prasad等[51]利用MICPs和f-MWCNTs复合材料构建手性电化学传感器用于手性识别蛋氨酸(Met)对映异构体。首先,将f-MWCNTs分散液用旋涂法修饰于电极表面形成均一的膜。然后,将该电极浸在Met、联苯胺和乙酸(pH=2.3) 混合液中,采用CV电聚合法在f-MWCNTs修饰的电极表面修饰上PANI膜,在电聚合过程中苯胺会被迅速氧化,电极上实际上为氧化苯胺印迹聚合物膜,将其用于检测水相、药物和血液中的L-Met,线性范围分别为11.7~206.3 ng/mL、11.7~197.4 ng/mL和11.8~152.3 ng/mL,检测限2.4~3.0ng/mL。f-MWCNTs和MIP的协同作用使电极具有电化学活性和机械强度高、背景电流低以及耐腐蚀、易修饰、经济、微型、可再生等优点。该传感器在临床医学分析方面有重要研究意义。
2016年,Gu等[52]对MIP-过氧化PPy(oPPy)-Au纳米颗粒(AuNPs)手性识别半胱氨酸(Cys)对映体进行研究。将MIP-AuNPs-oPPy应用于Cys对映异构体的手性电化学识别时,其识别效率优于MIP-oPPy,这归因于AuNPs可以有效吸附L-Cys模板并增加识别位点的数量,从而增强了识别效率。这项工作为基于AuNPs和导电聚合物复合膜的新型电化学手性传感器的构建提供了有益的参考。
3 基于多糖的手性识别电化学传感器
天然多糖指自然界中广泛存在的纤维素及其衍生物、甲壳素类及海藻酸、淀粉等高分子材料。天然多糖因具有高度的稳定性、无毒性、生物可降解性,而被用于药物缓释[53]和重金属离子去除[54]等众多领域。天然多糖具有手性环境,且其分子中含有大量—OH、—NH2、—COOH等活泼基团,可作为手性选择剂修饰于电极表面,与目标分子的官能团形成氢键作用,用于对手性化合物的识别和分离[55~59]。
3.1 基于β-环糊精的手性识别电化学传感器
环糊精(CD)是一种环状低聚糖,通常含有6~12个D-吡喃葡萄糖单元,研究得较多且具有重要实际意义的是含有6、7、8个葡萄糖单元的分子,分别称为α-、β-和γ-CD。CD中各葡萄糖单元均以1, 4-糖苷键结合成环。CD的特殊分子结构使它能够像酶一样提供一个疏水的结合部位而选择性地键合各种有机、无机和生物分子,形成主-客体配合物[60]。CD是通过“内识别”(主要通过范德华力、疏水作用力、色散力等弱相互作用力与客体形成“笼型”包络物)和“外识别”(主要是通过氢键力与客体形成“管道型”表面作用产物)两种方式,与各类客体分子通过分子间相互作用形成主-客体包合物[61],这也是CD实现手性识别的前提[62]。CD结构的另一个重要特点是手性,组成CD的每个葡萄糖单元有5个手性碳原子,可为客体分子提供良好的手性环境。图 4展示了CD的结构式及CD手性分子识别Trp对映体时的络合反应最优的络合状态。
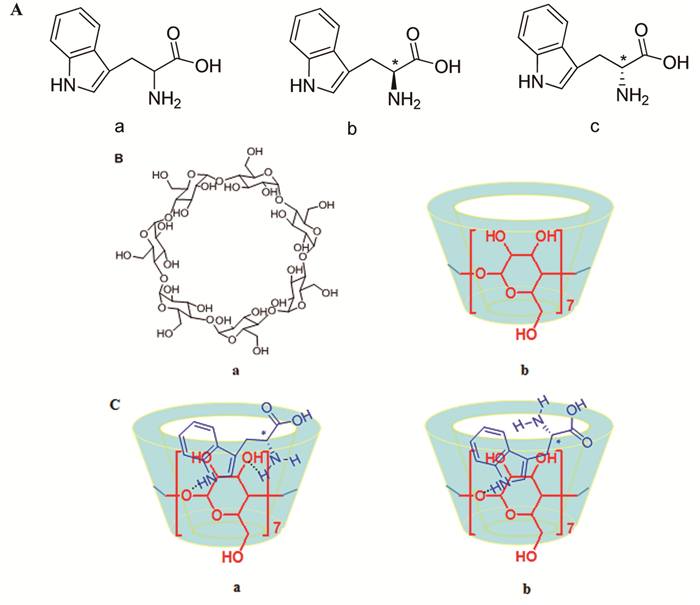 图 4
A:(a)Trp,(b)L-Trp,(c)D-Trp的结构式;B:CD的化学结构(a)和功能立体结构式(b);C:CD与色氨酸对映异构体络合后的最佳状态[63]
Figure 4.
(A) Chemical structure of (a) Trp, (b) L-Trp, and (c) D-Trp; (B) Chemical structure (a) and functional structural scheme (b) of CD; (C) Schematic diagram showing the optimal orientation of L-Trp (a) and D-Trp (b) on the basis of the highest degree of hydrogen bonding and inclusion complexation[63]
图 4
A:(a)Trp,(b)L-Trp,(c)D-Trp的结构式;B:CD的化学结构(a)和功能立体结构式(b);C:CD与色氨酸对映异构体络合后的最佳状态[63]
Figure 4.
(A) Chemical structure of (a) Trp, (b) L-Trp, and (c) D-Trp; (B) Chemical structure (a) and functional structural scheme (b) of CD; (C) Schematic diagram showing the optimal orientation of L-Trp (a) and D-Trp (b) on the basis of the highest degree of hydrogen bonding and inclusion complexation[63]
2014年,Feng等[64]研究了β-CD和氧化石墨烯(GO)复合材料电化学传感器手性识别Trp对映异构体。首先将CD接枝到碳二亚胺和羟基琥珀酰亚胺活化的GO上得到CD-GO复合物,还原得CD-还原石墨烯(GNs),用滴涂法将CD-GNs修饰在GCE电极上;作为比较,制备了GNs修饰的电极(GNs/GCE)。通过CV、EIS研究不同修饰电极的电化学行为。由于CD-Cu2+-L-Trp复合物中L-Trp的吲哚基进入到CD的空腔内,使结合力增强,所以CD-GNs/GCE对L-Trp响应信号强于D-Trp。该工作有助于理解基于配体交换的手性分子的高选择性,同时,它为药物分析和临床医学中各种手性对映异构体的识别提供了有益的参考。
Tao等[63]用CV法将L-Glu电化学聚合到GCE表面,然后将电极浸在含β-CD的溶液中过夜,制得β-CD/P-L-Glu/GCE修饰电极。该电极在最优条件下对Trp对映异构体的分离系数为2.30。β-CD掺杂复合物材料可提供电化学手性识别的新途径。例如,主体分子的空腔大小可以通过使用不同的CD来改变(α-、β-、γ-CD),因此可扩展到识别其他含芳香环的手性化合物。2015年,Tao等[65]将L-Glu通过CV法电沉积电极表面,再将二价铜修饰的β-CD(Cu2-β-CD)通过分子间氢键的作用自组装到P-L-Glu上,获得Cu2-β-CD/P-L-Glu/GCE。Cu2-β-CD的功能是防止空腔内的高能量水分子在络合时被释放出来,以及迫使Trp异构体从其较窄的开口进入空腔。在最佳温度和pH下,由于在空腔内的高能水分子倾向于与D-Trp的氨基形成氢键,从而实现Trp对映异构体的选择性识别。
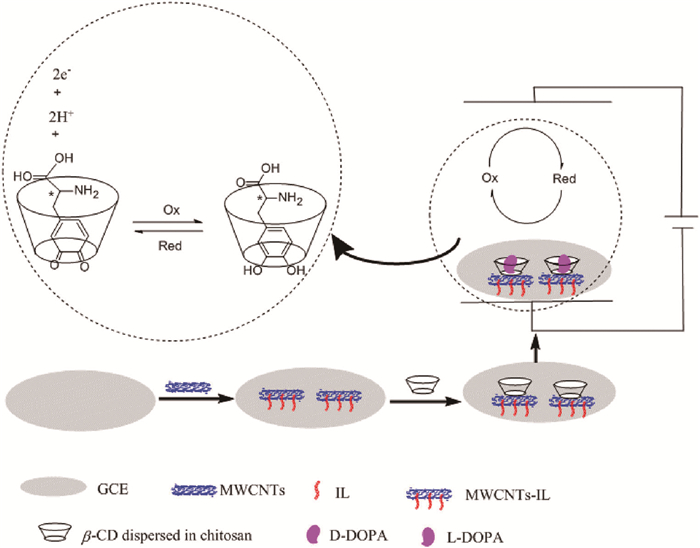
Chen等[66]将β-CD固定在f-MWCNTs-离子液体(IL)纳米复合材料的表面,构建了一种手性识别dopa对映体的灵敏的电化学方法。先后用滴涂法将离子液体(1-丁基-3-甲基咪唑六氟磷酸盐)与f-MWCNTs复合物溶液(f-MWCNTs-IL)和β-CD修饰于电极表面,得修饰电极(β-CD/f-MWCNTs-IL/GCE)(如图 5)。采用CV、EIS、SEM对β-CD/MWCNTs-IL的固定化过程进行了表征。该传感器通过DPV法对D-dopa进行检测,线性范围为4.0×10-3~4.0×10-9mol/L,检测限为1.2×10-9mol/L。
2016年,Xu等[67]成功合成β-CD/PtNPs/GNs复合纳米材料,并采用DPV法研究了Trp对映异构体在β-CD/PtNPs/GNs/GCE上的电化学行为,结果显示,L-和D-Trp的峰电流值有明显差异。在最优实验条件下,检测Trp的线性范围为5.0×10-5~5.0×10-3 mol/L,检测限为1.7×10-5(S/N=3)。该方法为检测Trp对映异构体提供了新的可行平台,检测性能优于Huang等[68]和Fan等[69]所报道的Trp对映异构体识别方法。2017年,Tao等[70]将由Cu2+修饰的β-CD自组装到氨性乙醇处理的CS(ae-CS)上,得到高度有序的自组装结构,并用于手性识别Trp对映体。结果表明,ae-CS/Cu-β-CD/GCE的识别效率(IL-Trp/ID-Trp=7.26) 是β-CD修饰电极的2.96倍。
3.2 基于壳聚糖手性识别的电化学传感器
CS是一种阳离子型天然多糖,分子内具有配位能力较强的氨基和羟基,可与目标分子氨基酸上的羧基和氨基形成氢键;此外,CS还具有天然的手性微环境、良好的成膜性,因此可将其用于电化学识别对映异构体。
2015年,Gu等[71]受DNA结构及大分子作用的启发来合成双螺旋结构的CS和磺化壳聚糖(SCS),用恒电位法电沉积CS或SCS于电极表面成膜识别Trp对映体。通过主客体间空间位阻作用而选择性形成分子间氢键的原理来识别D-Trp。对于SCS识别膜,磺化作用使得CS分子内氢键被破坏,从而加强了SCS与D-Trp分子间氢键作用,使得SCS识别效果要明显优于CS,其对L-Trp的响应信号强于D-Trp,而且SCS修饰电极可用于检测L-和D-Trp在外消旋混合物中的比例。通过电沉积CS和SCS电化学方法识别Trp异构体的研究,拓宽了天然多糖作为手性选择剂在电化学手性识别中的应用,在实际药品检测分析中有重大应用价值。
Ou等[72]通过CV逐步电沉积法将石墨烯量子点(GQDs)-CS复合膜修饰于电极表面,由于GQDs和CS两者之间的强的静电作用,使得在电极表面形成规整且均一的复合膜。该复合膜可用于电化学手性识别Trp对映体,CS在复合膜中提供手性微环境,同时GQDs能够放大电化学信号和提高识别效率。两者的协同作用使该复合膜成功有效地识别Trp对映体。和之前广泛报道的GQDs光致发光效应的研究相比,该工作为GQDs在电化学手性传感方面的应用开辟了新的道路。
2015年,Yu等[73]通过氧化热合法得到的MWCNTs-CS的悬浮液,并滴涂与电极表面,红外灯下干燥,得到MWCNTs-CS修饰电极,用于手性识别Trp对映异构体。L-和D-Trp的DPV响应不仅峰电流有很大的差异,而且峰电位也有明显差异;其中,外消旋混合物中L-Trp比例的增加使峰电位正向移动。该电极还可用于检测L-Trp在外消旋溶液中的百分比。
4 基于蛋白质手性识别的电化学传感器
蛋白质作为一种天然生物聚合物,具有独特的三维空间结构,有多个手性识别位点。在手性识别过程中,蛋白质结构中的疏水性口袋、沟槽或通道以及极性基团可与一些对映体形成非对映体,或者产生不同的氢键作用、静电作用、疏水作用等达到手性拆分的目的。蛋白质用于手性识别时要考虑缓冲溶液、pH和盐等的影响。
溶胶-凝胶方法是一种有效的固载蛋白质的方法,其中蛋白质的微观结构和化学环境没有改变,而且在空穴中可自由移动[74]。2012年,Wang等[75]研究了基于BSA修饰的电极用于青霉胺(Pen)对映体的立体选择性识别。他们采用Al2O3溶胶固载手性选择剂BSA,然后通过滴涂法修饰于电极表面,制得BSA修饰的电极。通过CV法对BSA的用量、反应时间、酸度和Pen对映体浓度对手性识别的影响进行了研究。结果显示,BSA对Pen对映体有不同的吸附能力,对D-Pen有更强的电化学响应信号。该研究促进了对蛋白质与手性药物的相互作用的了解,为手性药物的鉴定提供了参考。同年,Fu等[76]先将纳米金电沉积在电极表面,再将该电极浸在γ-球蛋白溶液中过夜,γ-球蛋白修饰到沉积纳米金的电极上,形成γ-球蛋白手性表面。他们通过CV和EIS法研究了手性表面的相关性质以及该手性表面对扁桃酸(MA)对映体的手性识别作用,并采用石英晶体微天平研究γ-球蛋白修饰手性表面吸附MA对映体后引起的频率变化情况,用AFM技术研究手性表面吸附MA对映体前后的形态特征。结果表明,该手性表面具有良好的电化学稳定性,在相同的实验条件下,由于R-MA的分子结构与γ-球蛋白更匹配,所以γ-球蛋白与R-MA的反应产生的电化学信号强于S-MA。并且,根据电流响应可以测定混合物中R-MA与S-MA对映体的比例。该方法具有电极修饰简单、检测快速灵敏的优点。
2013年,Zor等[77]报道了基于人血清白蛋白(HSA)/GO/3-氨丙基三乙氧基硅烷(APTES)修饰ITO来选择性识别D-和L-Trp。通过CV法研究了0.1~1.0 mmol/L D-和L-Trp在HAS/GO/APTES/ITO上的电化学行为,其峰电位分别为0.86和1.26 V,可以通过峰电位的差异同时识别D-和L-Trp,并且可以检测混合物中L-和D-Trp的比例。2015年,Kasinathan等[78]首先采用Humme’s法合成GO,还原后得到还原氧化石墨烯RGO,然后再与胶原蛋白(Col)组成纳米复合材料,将GO/Col和RGO/Col复合物分别滴在电极上获得GO/Col/GCE和RGO/Col/GCE修饰电极。他们通过CV法对扫描速率、电位范围、纳米复合材料的用量、pH、温度对手性识别的影响进行了研究;通过EIS法对GO/Col和RGO/Col复合物在电极表面的形态进行分析;最后通过CV和DPV法考察了修饰电极在手性分子识别方面的应用。该电极成功用于MA和Trp对映异构体的手性识别。
5 结语
由于手性化合物在医药、农药、食品添加剂等领域的广阔应用前景,发展实时在线、快速、简单、经济的手性识别技术显得尤为重要。电化学方法作为一重要发展的对象,可应用于临床检测、实际样品分析等,已引起人们的重点关注。本文从分子印迹技术、多糖和蛋白质三个角度对电化学方法手性识别的研究进展进行了综述。在MIP手性电化学传感器的研制中,要注重功能单体的选择,合成具有更广泛识别位点的功能单体,以提高印迹膜和目标分子之间的亲和性,能达到同时并准确手性识别多种分析物。MIP的制备和识别过程应更多集中在水相,更经济。总之,随着印迹技术的不断发展,MIP传感器将以其制备简单、针对性强、高选择性和重现性以及稳定性好、无污染等优点,在手性识别领域得到更广泛的应用。对于天然多糖手性选择剂,由于其自身多是柔性链状结构,且导电性不佳,在电极表面的检测信号较低,无法广泛应用于电分析化学领域。所以可将CS、β-CD进行改性(亲水性和空腔形状发生变化)或与其他材料进行复合(例如,碳纳米材料中的f-MWCNTs和GQDs),使得其性质更有利于专一性手性识别。由于蛋白质的空间结构、疏水效应等在手性识别过程中起着重要作用,所以蛋白质的活性结构不能发生变化,这就要求蛋白质在用于手性分析时要在适当的条件下进行。基于抗体-抗原相互作用的手性生物传感器[79, 80]由于特异性强、灵敏度高,是一个重要的发展趋势。借鉴生物识别体系,跟踪物理学、电子学的发展,相信不久的将来手性传感器会应用到更广泛的手性分子识别领域。
-
-
[1]
杜灿屏, 梁文平, 唐晋. 化学进展, 2002, 14(02): 156~158. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=hxjz200202011&dbname=CJFD&dbcode=CJFQ
-
[2]
于平, 岑沛霖, 励建荣. 中国生物工程杂志, 2001, 21(06): 89~94. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=swgj200106022&dbname=CJFD&dbcode=CJFQ
-
[3]
尹国, 刘振华, 曾姗姗等. 中国药物化学杂志, 2001, 11(01): 57~62. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=zgyh200101016&dbname=CJFD&dbcode=CJFQ
-
[4]
T Q Yan, C Orihuela. J. Chromatogr. A, 2007, 1156(1~2):220~227. http://www.sciencedirect.com/science/article/pii/S0021967307006280
-
[5]
Y Matsuoka, N Kanda, Y M Lee et al. J. Memb. Sci., 2006, 280(s 1~2):116~123. http://www.sciencedirect.com/science/article/pii/S0376738806000378
-
[6]
H Tabani, M Mahyari, A Sahragard et al. Electrophoresis, 2015, 36(2):305~311. http://europepmc.org/abstract/med/25262990
-
[7]
I M Mavridis, K Yannakopoulou. Int. J. Pharm., 2015, 492(1~2):275~290.
-
[8]
Y Ma, H Zhang, Z U Rahman et al. Electrophoresis, 2014, 35(19):2772~2777.
-
[9]
I Ali, M M Sanagi, H Y Aboul-Enein. Electrophoresis, 2014, 35(7):926~936.
-
[10]
M Dabrowska, M Starek. Food. Chem., 2014, 142(3):220~232. http://europepmc.org/abstract/med/24001835
-
[11]
E A Christodoulou. Curr. Org. Chem., 2010, 14(19):2337~2347.
-
[12]
L Challier, F Mavré, J Moreau et al. Anal. Chem., 2012, 84(12):5415~5420. http://europepmc.org/abstract/MED/22624981
-
[13]
H Y Xie, Z R Wang, Z F Fu. J. Pharm. Anal., 2014, 4(6):412~416.
-
[14]
Z Jie, Y Wang, L Yun et al. Anal.Chim.Acta, 2015, 868:73~79. http://europepmc.org/abstract/med/19720177
-
[15]
P Nowak, M Garnysz, M P Mitoraj et al. J. Chromatogr. A, 2015, 1377:106~113.
-
[16]
L Libo, L Xia, L Quan et al. Talanta, 2015, 142:28~34.
-
[17]
G Neumajer, T Sohajda, A Darcsi et al. J. Pharm. Biomed. Anal, 2012, 62(62):42~47. http://www.sciencedirect.com/science/article/pii/S0731708511007394
-
[18]
Z I Szabó, G Tóth, G V lgyi et al. J. Pharm. Biomed. Anal., 2015, 30(4):359~360.
-
[19]
I Ali, D R Sahoo, Z A Alothman et al. J. Chromatogr. A, 2015, 1406:201~209.
-
[20]
T Sheng, B Qin, C Wei et al. J. Chromatogr. A, 2016, 1440:112~122.
-
[21]
C Lin, J Fan, W N Liu et al. J. Pharm. Biomed. Anal., 2014, 98(10):221~227.
-
[22]
V Kannappan, S S Mannemala. J. Pharm. Biomed. Anal., 2015, 120(2):270~278. http://europepmc.org/abstract/MED/26760239
-
[23]
陈巧, 西南大学硕士学位论文, 2012.
-
[24]
M H Xu, J Lin, Q S Hu et al. J. Am. Chem. Soc., 2002, 124(47):14239~14246. http://europepmc.org/abstract/MED/12440923
-
[25]
H L Liu, P Qian, Y D Wu et al. Angew. Chem. Int. Ed., 2010, 49(3):612~616. doi: 10.1002/ange.200904889/asset/supinfo/ange_200904889_sm_miscellaneous_information.pdf?v=1&s=f0437b661a7a9fa3c5cf086d150e805455303f85
-
[26]
S O Fakayode, I M Swamidoss, M A Busch et al. Talanta, 2005, 65(4):838~845.
-
[27]
C Guo, R D Shah, R K Dukor et al. Anal. Chem., 2004, 76(76):6956~6966. http://europepmc.org/abstract/med/15571347
-
[28]
W A Tao, R G Cooks. Anal. Chem., 2003, (1):25A~31A.
-
[29]
A Kühnle, T R Linderoth, B Hammer et al. Nature, 2002, 415(6874):891~893.
-
[30]
W S Kim, H Y Lee, T Kawai et al. Sens. Actuat. B, 2008, 129(1):126~133.
-
[31]
陈婷, 万立骏, 中国科学: 2009, (10): 1102~1114. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=jbxk200910008&dbname=CJFD&dbcode=CJFQ
-
[32]
尹秀丽, 复旦大学博士学位论文, 2005.
-
[33]
J Huang, Z Wei, J Chen et al. Sens. Actuat. B, 2008, 134(2):573~578. http://www.sciencedirect.com/science/article/pii/S0925400508003134
-
[34]
E Granot, R Tel Vered, O Lioubashevski et al. Adv. Funct. Mater., 2008, 18(3):478~484.
-
[35]
刘志航, 宦双燕, 浣石等. 化学传感器, 2005, 25(4): 1~8.
-
[36]
苏立强, 宁振鑫, 于思明等. 理化检验(化学分册), 2013, (01): 11~14. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=lhjh201301007&dbname=CJFD&dbcode=CJFQ
-
[37]
J Damen, D C Neckers. Tetrahed. Lett., 1980, 21(20):1913~1916. http://www.sciencedirect.com/science/article/pii/S0040403900936426
-
[38]
L Andersson, B Sellergren, K Mosbach. Tetrahed. Lett., 1984, 25(45):5211~5214. http://www.sciencedirect.com/science/article/pii/S0040403901815665
-
[39]
S Fireman-Shoresh, I Turyan, D Mandler et al. Langmuir, 2005, 21(17):7842~7847.
-
[40]
S Sekine, Y Watanabe, Y Yoshimi et al. Sens. Actuat. B, 2007, 127(2):512~517.
-
[41]
H Liao, Z Zhang, L Nie et al. J. Biochem. Bioph. Meth., 2004, 59(1):75~87. http://europepmc.org/abstract/MED/15134909
-
[42]
B B Prasad, S Srivastava, K Tiwari et al. Mater. Sci. Eng:C, 2009, 29(6):1781~1789. http://www.sciencedirect.com/science/article/pii/S0928493109000460
-
[43]
B B Prasad, D Kumar, R Madhuri et al. Biosens. Bioelectron., 2011, 28(1):117~126. http://www.sciencedirect.com/science/article/pii/S0956566311004362
-
[44]
B B Prasad, I Pandey. Sens. Actuat. B, 2013, 186(18):407~416.
-
[45]
T Sajini, B Mathew. J. Surf. Sci. Technol., 2014, 30(3~4):201~208.
-
[46]
B C Iacob, E Bodoki, A Florea et al. Anal. Chem., 2015, 87(5):2755~2763. http://europepmc.org/abstract/med/25630982
-
[47]
倪珺华, 常州大学硕士学位论文, 2011.
-
[48]
B Deore, Z Chen, T Nagaoka. Anal. Chem., 2000, 72(17):3989~3994. http://europepmc.org/abstract/med/10994955
-
[49]
V Syritski, J Reut, A Menaker et al. Electrochim. Acta, 2008, 53(6):2729~2736. http://www.sciencedirect.com/science/article/pii/S0013468607012947
-
[50]
I Basozabal, A Gómez-Caballero, N Unceta et al. Electrochim. Acta, 2011, 58(1):729~735.
-
[51]
B B Prasad, I Pandey, A Srivastava et al. Sens. Actuat. B, 2013, 176(6):863~874.
-
[52]
J Gu, H Dai, Y Kong et al. Syn. Met., 2016, 222:137~143.
-
[53]
A Rubinstein. Drug Dev. Res., 2000, 50(3~4):435~439.
-
[54]
B C Son, K Park, S H Song et al. Korean J. Chem. Eng., 2004, 21(6):1168~1172. doi: 10.1007/BF02719489
-
[55]
S Wang, Y Dai, J Wu et al. J. Chromatogr. A, 2013, 1277:84~92. http://www.sciencedirect.com/science/article/pii/S0021967312019322
-
[56]
T Jian, Y Lu, Y Wang et al. Talanta, 2014, 128:460~465. http://www.sciencedirect.com/science/article/pii/S187439191400102X
-
[57]
M Monier, D M Ayad, Y Wei et al. Biochem. Eng. J, 2010, 51(3):140~146. http://www.sciencedirect.com/science/article/pii/S1369703X10001634
-
[58]
J H Kim, J Jegal, J H Kim et al. J. Appl. Polym. Sci, 2003, 89(11):3046~3051.
-
[59]
Y Wang, S Zhu, Y Liao et al. J. Polym. Res., 2014, 21(12):1~7.
-
[60]
F Ortega-Caballero, C Rousseau, B Christensen et al. J. Am. Chem. Soc., 2005, 127(10):3238~3239. http://europepmc.org/abstract/MED/15755115
-
[61]
陈丽娟, 杨明星, 林深. 合成化学, 2002, 10(03): 205~210. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=hchx200203005&dbname=CJFD&dbcode=CJFQ
-
[62]
韩翠平, 华中师范大学博士学位论文, 2012.
-
[63]
Y Tao, J Dai, Y Kong et al. Anal. Chem., 2014, 86(5):2633~2639. http://europepmc.org/abstract/med/24484527
-
[64]
W Feng, C Liu, S Lu et al. Microchim. Acta, 2014, 181(5~6):501~509.
-
[65]
Y Tao, X Gu, L Deng et al. J. Phys. Chem. C, 2015, 119(15):8183~8190.
-
[66]
Y Chen, Y Huang, D Guo et al. J. Solid. State. Electrochem., 2014, 18(12):3463~3469.
-
[67]
J Xu, Q Wang, C Xuan et al. Electroanalysis, 2016, 28(4):868~873. doi: 10.1002/elan.201500548/pdf
-
[68]
K J Huang, C X Xu, W Z Xie et al. Colloid. Surf. B, 2009, 74(1):167~171.
-
[69]
Y Fan, J H Liu, H T Lu et al. Microchim. Acta, 2011, 173(1~2):241~247.
-
[70]
Y Tao, X Gu, B Yang et al. Anal. Chem., 2017, 89(3):1900~1906.
-
[71]
X Gu, Y Tao, Y Pan et al. Anal. Chem., 2015, 87(18):9481~9486.
-
[72]
J Ou, Y Tao, J Xue et al. Electrochem. Commun., 2015, 57:5~9.
-
[73]
L Y Yu, Q Liu, X W Wu et al. RSC Adv., 2015, 5(119):98020~98025.
-
[74]
张宇, 陈年友. 高等学校化学学报, 2000, 21(05): 675~680. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=gdxh200005003&dbname=CJFD&dbcode=CJFQ
-
[75]
Y Wang, Q Han, Q Zhang et al. J. Solid State Electrochem., 2013, 17(3):627~633. doi: 10.1007/s10008-012-1859-4
-
[76]
Y Fu, Q Chen, J Zhou et al. Anal. Biochem., 2012, 421(1):103~107.
-
[77]
E Zor, I H Patir, H Bingol et al. Biosens. Bioelectron., 2013, 42:321~325. http://europepmc.org/abstract/MED/23208105
-
[78]
B Kasinathan, R M Zawawi, N L Hong. J. Appl. Electrochem., 2015, 45(10):1085~1099.
-
[79]
M Trojanowicz. Electrochem. Commun., 2014, 38(1):47~52.
-
[80]
B Wei, N Liu, J Zhang et al. Anal. Chem., 2015, 87(4):2058~2062.
-
[1]
-


图 2 在100mmol/L pH 7.0的PBS溶液中(10min,浓度为1μg/mL)R型(黑色线)和S型(红色线)的β-受体阻滞剂的DPV扫描曲线[46]
Figure 2 Normalized DPVs obtained in 100mmol/L phosphate buffer solution (pH=7.0) after rebinding (10min, 1μg/mL) of R(+)-(black line) and S(-)-(red line)[46]
R(+)ATNL-分子印迹聚合物修饰电极,A:AYNL,B:PRNL,C:ALPRNL,D:OXPRNL;R(+)PRNL-分子印迹聚合物修饰电极,E:PRNL,F:ALPRNL,G:AYNL,H:OXPRNL


图 4 A:(a)Trp,(b)L-Trp,(c)D-Trp的结构式;B:CD的化学结构(a)和功能立体结构式(b);C:CD与色氨酸对映异构体络合后的最佳状态[63]
Figure 4 (A) Chemical structure of (a) Trp, (b) L-Trp, and (c) D-Trp; (B) Chemical structure (a) and functional structural scheme (b) of CD; (C) Schematic diagram showing the optimal orientation of L-Trp (a) and D-Trp (b) on the basis of the highest degree of hydrogen bonding and inclusion complexation[63]
-


 下载:
下载:




 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 0
- HTML全文浏览量: 0
 下载:
下载:
