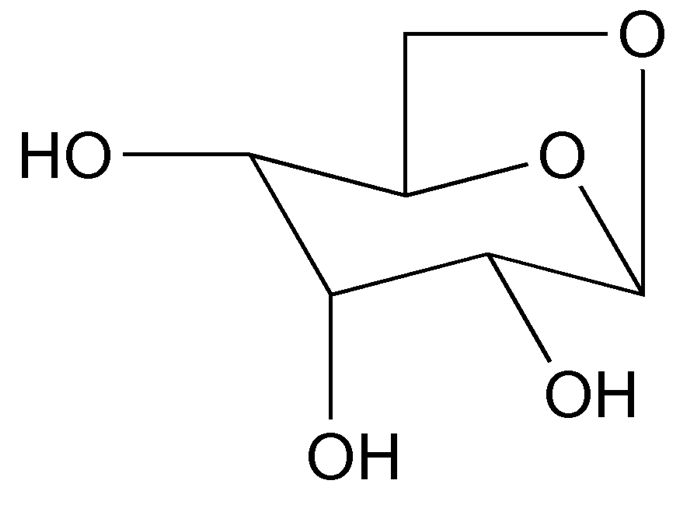
 图 图式1
左旋葡聚糖结构式
Figure 图式1.
The structural formula of levoglucosan
图 图式1
左旋葡聚糖结构式
Figure 图式1.
The structural formula of levoglucosan
引用本文:
李文惠, 武红丽, 黄婷, 邓茹, 曹飞, 韦萍. 左旋葡聚糖的制备与在生物技术领域的应用[J]. 化学通报,
2017, 80(3): 251-259.

Citation: Li Wenhui, Wu Hongli, Huang Ting, Deng Ru, Cao Fei, Wei Ping. Preparation of Levoglucosan and Its Applications in Biotechnology[J]. Chemistry, 2017, 80(3): 251-259.
Citation: Li Wenhui, Wu Hongli, Huang Ting, Deng Ru, Cao Fei, Wei Ping. Preparation of Levoglucosan and Its Applications in Biotechnology[J]. Chemistry, 2017, 80(3): 251-259.
左旋葡聚糖的制备与在生物技术领域的应用
English
Preparation of Levoglucosan and Its Applications in Biotechnology
Abstract:
Levoglucosan is a potential source of sugar, it can be hydrolyzed into glucose and then used for microbial fermentation, or be directly metabolized through levoglucosan kinase or levoglucosan dehydrogenase in some fungi and bacteria. It showed a good application prospect in the fermentation field. In addition, levoglucosan can also be used as a carbon source to produce bio-ethanol, succinic acid and so on. At present, biomass pyrolysis is the common-use preparation method of levoglucosan. However, this method has the disadvantages of high energy consumption, low yield and difficult to extract the product. In order to improve the yield of levoglucosan, we should strive to develop a method of decomposing cellulose in solvent system. Future researches should focus on the development new processes, new methods, and new strains for microbial metabolism of levoglucosan, which could lay the groundwork for the efficient use of levoglucosan.
-
Key words:
- Levoglucosan
- / Biomass
- / Microbial fermentation
- / Carbon source
-
左旋葡聚糖 (Levoglucosan,LGA) 是生物质燃烧 (主要是纤维素热解) 生成的一种脱水糖[1],经常存在于土壤、气溶胶、冰雪、甚至人类的尿液中,可被用作监测PM2.5的分子示踪物[2~7]。Jordan等[8]报道了一种以LGA作为大气中稳定的示踪物来量化木材燃烧的烟雾对大气的污染程度的方法。除了LGA,在气溶胶和土壤样本中也发现了一些其他脱水糖,如甘露聚糖、半乳聚糖、左旋半乳聚糖、左旋甘露聚糖和聚纤维二糖,但它们的含量都不如LGA高[2, 3, 5, 6]。据估计,全球每年生物质燃烧释放80亿吨碳[9]。通过对生物质燃烧产生的烟雾进行表征发现,脱水糖是第二大量的有机碳组分 (仅次于酸类物质),含量为24mg/g有机碳,进而可以估算脱水糖年排放量为1.8亿吨[10]。这些脱水糖成为目前全球碳循环中具有重大意义但未被充分开发的一部分。
目前,大部分LGA可通过木质纤维素类生物质热解而制备。与葡萄糖相比,LGA仅仅是在1, 6-位脱去一分子水,容易通过水解反应再形成葡萄糖[11]。由于木质纤维素类生物质广泛易得,所以将其热解得到的脱水糖转化为生物燃料或者其他有价值的化学品也展现出巨大的商业应用潜力。因此,LGA可以作为新的糖源应用于生物基平台化合物制备和生物技术发酵领域[12~19]。
1 LGA的物化性质
LGA,化学名:1, 6-脱水-β-D-吡喃葡萄糖,CAS号498-07-7;分子式为C6H10O5;分子量162.14;熔点183℃;结构式见图式1。由于含有多个羟基,LGA是一种极性化合物,易溶于水,且挥发性低。
图 图式1
左旋葡聚糖结构式
Figure 图式1.
The structural formula of levoglucosan
2 LGA的制备
2.1 生物质热解途径制备LGA
生物质在250~550 ℃之间有氧分解为热解焦、液体和气体三种产物,这个过程叫做热解。当热解温度在400~550 ℃、生物质颗粒很细小 (直径小于2毫米) 并且升温速率很高时,热解产生的生物油的量达到最大 (66%),这个过程被称为快速热解。当热解温度在250~350 ℃,生物质颗粒较大 (直径达到厘米级别) 时,热解产生的热解焦的量会增加,这个过程被称为慢速热解或炭化。热解是所有生物质燃烧和气化的第一步化学反应[20]。其中液态产物为热解油或者生物油,可经反应器单级或多级 (阶段式) 冷凝器冷凝后浓缩收集得到 (如图 1所示)[20]。在三种形式的产物中,生物油最有价值,因为它能进一步转化为各种各样的化学产品[1]。生物油是数百种化合物组成的复杂的混合物[21, 22]。除了醛类、酮类、酸类、呋喃类和酚醛树脂类,生物油也包含一些脱水糖,主要是LGA[23]。
 图 1
生物质热解过程示意图
Figure 1.
Biomass pyrolysis process diagram
图 1
生物质热解过程示意图
Figure 1.
Biomass pyrolysis process diagram
2.2 非热解途径生物质制备LGA
Cao等[55]报道了纤维素在极性非质子溶剂中分解,以含水量2.7(wt)%的THF作溶剂,纤维素投加量1(wt)%,7.5mmol/L硫酸催化,190℃反应下30min,LGA产率为33%。
Ohara等[56]报道了用大孔树脂-15固体酸催化剂催化葡萄糖选择性的脱水合成LGA,以DMF为溶剂时,LGA产率达32%。
2.3 LGA的分离提取
为了降低生物油的化学复杂度,可用水对其进行提取。水提取物包含了生物油中的极性化合物,主要为脱水糖,如此就将亲水性的LGA与非极性化合物分开[57]。生物油含水量多少不仅取决于热解工艺条件,也取决于生物质材料的含水量。木材生物油的含水量为15%~30%[58],干草和秸秆生物油的含水量为39%~51%[59],米糠生物油的含水量为28%[60]。当水含量超过一个特定的界限 (30%~45%之间[61]) 时,生物油就会分为两相:一相是富含糖并伴有其他亲水化合物的水相;一相是主要包含木质素派生的低极性化合物[62]的非水相。
许多研究者已经致力于优化从生物油中分离出含高浓度LGA的水相的工艺参数。Bennett等[11]优化了从欧洲赤松生物油中提取出含高浓度LGA的水相的条件:向生物油中加入62(wt)%的水,在34℃条件下保持22min,可得到LGA浓度为87g/L的水相。Chan等[63]采用水与生物油质量比1 :1进行分离,LGA产率为4.98%。Wang等[64]将水与火炬松生物油体积比1 :1混匀,室温下静置48h,能达到完全相分离。Zhang等[65]采用水与生物油1 :1在50℃条件下接触20min,得到LGA浓度为4.1(wt)%的水相。以上研究结果表明,生物油的相分离效果不仅取决于生产生物油的生物质材料,还取决于水与生物油的配比、提取温度及接触时间。
综上所述,虽然有很多生物质热解制备LGA的研究,但是它们都存在以下一些问题:(1) 反应温度高 (250~550℃),能耗大;(2) 多数数据来自于微型热解反应器,大量制备的重现性有待考察;(3) LGA存在于生物油中,而生物油成分复杂,提取困难、收率低。相比而言,采用溶剂体系中分解纤维素制备LGA具有重大的应用价值,反应温度低,产物容易提取。
2.1.1 LGA的热解制备机理
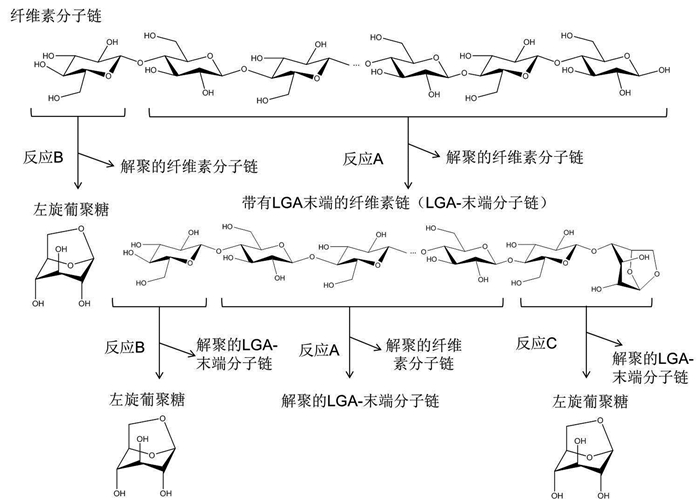
Hosoya等[24]提出了LGA可能的形成机制,如图式2所示。第一种是反应A,内部糖基的反应,生成两个解聚链,一个是带有LGA末端的纤维素链,另一个是更短的纤维素链;第二种是反应B,非还原端的反应,生成LGA和少了一个糖聚合度的纤维素链;第三种是反应C,反应A产生的LGA末端相邻的葡萄糖基发生反应,生成LGA和一个新的LGA末端。研究结果表明,LGA主要是由反应A生成的LGA末端进行反应C得到。
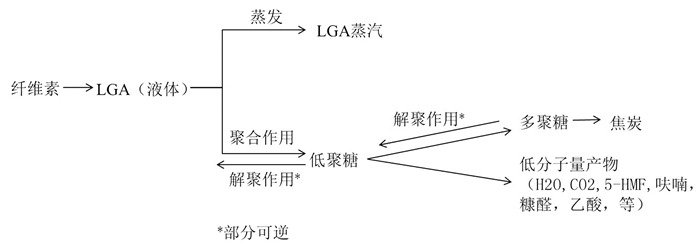
对于LGA在纤维素热解中的形成机制、热分解机制以及LGA合成动力学、密度泛函理论计算、热力学解释等,许多学者进行了深入研究[25~30]。Bai等[30]根据其研究结果提出了一种纤维素类生物质热解路径,如图 2所示。纤维素热解首先生成含LGA的生物油,然后生物油中的LGA发生了两个竞争性反应:蒸发以及聚合为低聚糖。低聚糖进一步聚合为多糖,同时脱水生成低分子量产物。多糖可进一步脱水生成同样的低分子量产物和热解焦,或部分重新解聚为低聚糖。
2.1.2 LGA热解制备工艺优化
关于纤维素热解制备LGA,国内外已经有很多学者做了大量研究,工艺改进主要针对纤维素预处理、改变反应体系压力、反应器优化等方面[31~45],表 1总结并分析了其中具有显著价值的研究结果。
 表 1
由生物质热解制备左旋葡聚糖的部分文献报道
Table 1.
Part of references about the preparation of levoglucosan by cellulose pyrolysis
表 1
由生物质热解制备左旋葡聚糖的部分文献报道
Table 1.
Part of references about the preparation of levoglucosan by cellulose pyrolysis
原料 反应器类型 预处理及催化 LGA产率 参考文献 玉米芯 CDS 5200型微型热解反应器 在甘油中微波预处理 (150W,1min) 38% [31] 纤维素 固定床反应器 固体酸催化 22.1% [39] 微晶纤维素 小型管式反应器 380℃、25MPa条件下,超临界水预处理0.4s 低聚糖产率42%,其中LGA浓度为8mg/mL [32] 黄杉木 CDS 5000型微型热解反应器 稀硝酸酸洗预处理后加入0.05%硫酸作催化剂 没有定量 [34] 杨木 流化床反应器 90℃下用5%硫酸酸洗,之后水洗至pH为6.6 30.42% [33] 松木 20℃单级冷凝器 去矿物质化 15.8% [35] 杨木 实验室规模螺旋钻反应器 高压热水预处理 10.56% [36] 木材 石英管反应器 3mol/L硫酸预处理 8.5% [37] 甘蔗渣 微型裂解炉 稀酸预处理 40.5% [38] 微晶纤维素 快速冷却、连续给料裂解炉,减压热解 (5kPa) 70.1%(分析值) [41] 纤维素 真空条件下 (低于1mmHg),特制玻璃热解反应器 50%~55% [40] 纤维素 微型热解反应器 58.78% [43] 纤维素 CDS 5200型微型热解反应器 56% [44] 松木 反应器改造:在冷凝前的蒸汽流中注入水喷雾 稀酸预处理 热解油的16.43% [45] 玉米淀粉 管式炉热解反应器,减压热解 (600Pa) 56%(分析值) [42] Pictet等[46]最早开展了通过热解纤维素制备LGA的研究。他们在真空条件下热解纤维素,得到了两相物质:一相是含LGA的水相,占32(wt)%;一相是黄色糊状物,占44(wt)%。尽管生物质快速热解可以得到75(wt)%的粗生物油,但是只有10(wt)%~20(wt)%的纤维素转化为LGA,而余下的则转化成为没有经济价值的小分子物质和热解焦。
有研究者发现向生物质中添加少量的硫酸可以使LGA的产率提高两倍[47],然而,大多数情况下LGA的产率都很低。许多研究者探索了LGA产率低的原因[48~53],例如,Lédé[51]认为生物质自身携带的少量碱金属 (比如钠和钾) 会启动碳链裂解反应,从而降低热解糖的产量;也有报道称生物质自身携带的碱金属会形成稳定的盐而引起钝化,减少了LGA的总产量[54]。针对这一情况,对生物质进行酸洗可以降低碱金属的影响[35]。此外,热解过程中纤维素与生物质中的其他组分发生的不良反应也会降低LGA的产率。
Zheng等[31]将生物质在甘油中进行微波预处理,可以实现快速加热和特定分子活化以促进生物质的去木质素作用和去矿化作用,使得LGA产率提高至38%(未预处理的产率为0.2%)。Tolonen等[32]将微晶纤维素在380℃、25MPa超临界水中处理0.4s,热解后低聚糖的产率提高至42%,其中LGA浓度为8mg/mL。Piskorz等[33]报道了在90℃条件下将杨木用5%硫酸酸洗后再水洗至pH为6.6,LGA产率可提高至30.42%。与之相比,Pecha等[34]报道了用稀硝酸酸洗预处理并且加入0.05(wt)%硫酸作催化剂,结果表明,酸洗和酸催化相结合比只酸洗 (Piskorz等[33]) 的LGA产率提高了85%。Oudenhoven等[35]对松木进行去矿物质化预处理,生物油产率可达66%,其中LGA占24%。Le Roux等[36]对杨木进行高压热水预处理,LGA产率从6.87%提高至10.56%。Kumagai等[37]发现生物质在室温下用3mol/L硫酸搅拌预处理,可以使纤维素、半纤维素、木质素中的C—O键断裂,生物油产率从30.1%提高至46.8%,LGA产率从1%提高至8.5%。Jiang等[38]研究发现,采用稀酸预处理的甘蔗渣在微型裂解炉中快速热解制备LGA的产率可达40.5%,效果比水洗 (29.1%) 和未预处理 (12.84%) 的都要好。Xia等[39]研究了Zn/ZSM-5和FePO4这两种固体酸催化对纤维素热解的影响,发现固体酸催化会降低LGA的产率,提高左旋葡萄糖酮的产率。尽管已经证实酸洗预处理和加入稀酸催化相结合是提高LGA产率的有效途径,但是要达到LGA的可利用产率仍面临着挑战。
有研究表明,减压热解对LGA的形成也是有利的。Kwon等[40]在真空 (低于1mmHg) 条件下进行热解,LAG产率达50%~55%;随后,他们在一个实验室规模的热解反应器中减压 (5kPa) 条件下纤维素热解,LGA产率达到70.1%(气相色谱检测值)[41]。Yang等[42]报道了玉米淀粉在管式炉热解反应器中、600Pa下热解,LGA产率为56%(气相色谱检测值)。
也有很多学者尝试了生物质在不同的反应器中进行热解。例如,Kwon等[41]研究了微晶纤维素在快速冷却、连续给料的裂解炉中减压热解,LGA产率达到70.1%(气相色谱检测值)。Patwardhan等[43]在微型热解反应器中热解纤维素,LGA产率达58%。Bai等[44]在CDS 5200型微型热解反应器中热解纤维素,当热解样本为59μg时,LGA产率高达56%。Li等[45]在热解反应器的冷凝器前加了喷雾装置,可以在冷凝前的蒸汽流中注入水喷雾,与只用稀酸预处理相比,在蒸汽流中注入水喷雾和稀酸预处理相结合,使生物油中LGA产率显著提高了30.7%(LGA占生物油的比例提高至16.43%)。
3 LGA的生物转化途径
相对于葡萄糖、蔗糖、果糖这些天然碳源,LGA在自然界中较稀少,但作为自然界中生物质燃烧释放出来的碳组分,其储量却是极其丰富的。因此,LGA是一种极具潜力的待开发的发酵碳源和能源。LGA能直接被微生物代谢或者经弱酸水解为葡萄糖后被微生物代谢[66, 67]。就目前研究现状来看,LGA的生物利用途径有3条:(1) 加酸水解为葡萄糖后用于微生物发酵[68~71];(2) 经左旋葡聚糖激酶 (Levoglucosan kinase,LGK)[72~74, 17, 18]转化为葡萄糖-6-磷酸,进入糖酵解途径[73],这条路径不仅存在于真菌中,并作为模块已经在细菌 (例如大肠杆菌) 中实现功能表达[74];(3) 左旋葡聚糖脱氢酶 (Levoglucosan dehydrogenase,LGDH) 途径,即通过一个依赖烟酰胺腺嘌呤二核苷酸 (NAD+) 的脱氢酶催化将LGA转化为3-酮-左旋葡聚糖,进而经过3-酮-左旋葡聚糖,3-酮-葡萄糖,最终生成D-葡萄糖[75]。关于LGK和LGDH具体的作用机制以及左旋葡聚糖膜转运蛋白的鉴定还在探索中[72]。其中途径 (1) 为LGA的间接生物转化,途径 (2)、(3) 为LGA的直接生物转化。下面是对这三种途径的举例说明。
3.1 LGA的间接生物转化
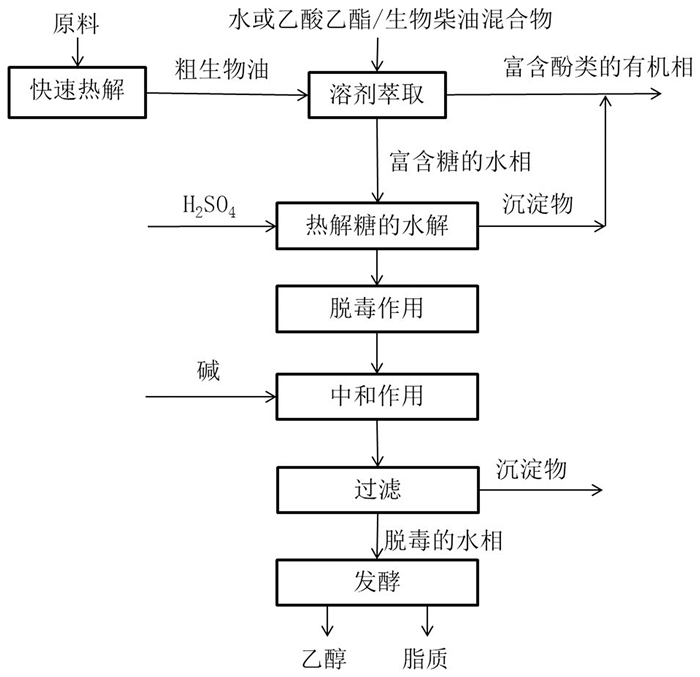
Lian等[68]将生物质热解产生的脱水糖水解为葡萄糖,并用于酵母菌发酵产乙醇和油脂。Helle等[77]研究了由生物质热解油获得LGA并将其水解为葡萄糖的整个过程。如图 3所示[76],首先以溶剂萃取分离热解糖和酚类化合物,然后用硫酸催化水解LGA为葡萄糖。毒理学研究表明,抑制酵母菌生长的主要是酚类化合物和酸。所以,用氢氧化钡中和硫酸和生物油中产生的羧酸,然后将糖浓缩液用活性炭脱毒。最后,由酵母菌来利用所得的富含葡萄糖的水溶液进行发酵。
3.2 LGA的直接生物转化
3.2.2 LGDH途径
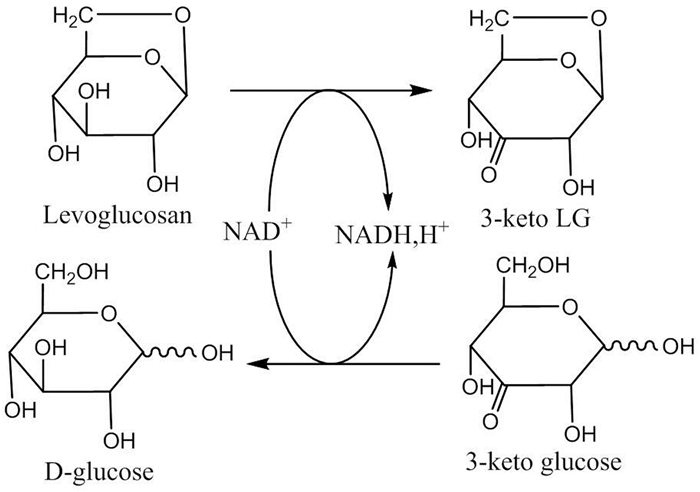
Nakahara等[75]从土壤中分离出一种可直接利用LGA的细菌,它属于节细菌属Arthrobacter,暂时命名为Arthrobacter sp. 1-552。该菌体中含有LGDH,可以催化LGA脱氢生成3-酮-左旋葡聚糖,3-酮-左旋葡聚糖继而转化为3-酮-葡萄糖,最终转化为D-葡萄糖。如图式4所示,通过纯化、表征该酶,验证了葡萄糖形成的途径。此途径不同于酵母和真菌中LGA的转化途径。
3.2.1 LGK途径
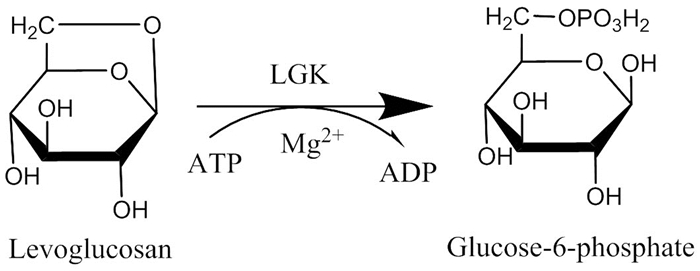
在酵母菌和丝状真菌中,在LGK的催化作用下,LGA的1, 6-酐键断裂,同时在镁离子和三磷酸腺苷 (ATP) 的存在下被LGK磷酸化,转化为葡萄糖-6-磷酸 (如图式3所示)[73, 78]。葡萄糖-6-磷酸可转入三羧酸循环 (TCA) 中,从而完全利用LGA。显然,在真菌中LGA的一步直接生物转化非常吸引人[18]。
原本推测LGK会是一个对LGA有亲和力的选择性功能蛋白,然而所有鉴别出的LGK对LGA的亲和力都很低,米氏常数 (Km) 高达48~102 mmol/L[79, 80]。这严重阻碍了LGA的直接转化。Bacik等[72]研究了LGK的晶体结构,结果显示,其结构中包含两个金属结合位点和一个LGA替换位点。这种不寻常的LGA结合方式造成LGK对LGA的结合力降低,需要进一步的酶改造来提高对LGA的结合力。
4 LGA在生物技术方面的应用
生物质作为一种引人注目的可再生资源,可以生产生物燃料及化学品。但是,木质纤维素固有的抗降解屏障,使其难以高效地解聚为干净的、可发酵的糖[81]。而生物质快速热解可以使很大一部分生物质转化为脱水糖[82]。脱水糖化学转化方面的应用研究也有很多。例如,Hu等[12]研究了多种溶剂中酸催化下将LGA转化为生物基平台化合物;Galletti等[83]报道了在乙腈和离子液体中的LGA酶催化酰化反应,得到一些具有表面活性剂性能的潜在产品;Kumar等[84]通过介孔材料催化LGA转化为糠醛。LGA还是合成具有生物活性 (比如抗艾滋病毒活性和抗凝血剂活性) 的立体规则多糖的重要原材料[85]。在这里,我们将集中阐述LGA在生物技术方面的应用。通过引入脱水糖代谢途径,目前已经分离出了可利用LGA的微生物,并且应用于发酵生产有价值的产物[86]。
4.1 LGA的间接利用--乙醇发酵
近几年,作为一种新的替代能源,纤维素乙醇广受期待[87, 88]。目前已经有一些关于热解油间接发酵生产乙醇的研究[11, 63, 67~69, 71, 89~91]。首先利用酸催化将脱水糖 (主要是LGA) 水解为葡萄糖,然后通过微生物利用所得的葡萄糖来发酵。
Prosen等[86]评估了一些真菌和酵母菌菌株利用含LGA的热解油生产乙醇的潜力。他们发现一些真菌和酵母菌菌株既能利用活性炭脱毒后的热解油,也能利用热解油的酸水解液。然而,利用酸水解液所得的乙醇产率比利用活性炭脱毒的热解油所得的乙醇产率要高,这说明加酸水解可能将LGA转化为葡萄糖--乙醇发酵的首选基质。此后,利用热解油的水解液 (含葡萄糖) 间接发酵产生物燃料 (尤其是生物乙醇和脂质) 的研究越来越多 (部分报道示于表 2),以生物质热解为基础生产乙醇的生物炼制过程展现出光明的前途。
表 2
左旋葡聚糖的间接利用 (乙醇发酵) 的部分相关文献
Table 2.
Part of references about the indirect use of levoglucosan (alcoholic fermentation)
提高乙醇产率的策略 发酵微生物 最高乙醇浓度/(g/L) 乙醇产量/(g/g葡萄糖) 参考文献 H2SO4水解+Ca (OH)2中和+硅藻土震荡 S.cerevisiae CCTCC 2.399 16.1 0.45 [71] Pichia sp. YZ-1 15.1 0.42 [71] H2O萃取+H2SO4水解+NaOH中和+高接种浓度+微需氧条件 S.cerevisiae T2 未提及 0.46 [11] H2O萃取+H2SO4水解+Ca (OH)2过度中和+有机溶剂萃取+自适应进化 S.cerevisiae T2 未提及 0.45 [63] 生物柴油或乙酸乙酯萃取+H2SO4水解+Ba (OH)2中和+活性炭 S.cerevisiae ATCC 200062 32~35 0.47 [68] Yu等[71]为了高效地利用废弃棉花热解油的酸水解液生产乙醇,在所考察的10种脱毒策略中,中和作用与硅藻土振荡相结合处理的水解液几乎可完全被酿酒酵母Saccharomyces cerevisiae2.399和毕赤酵母Pichia sp. YZ-1利用,其中酿酒酵母发酵所得乙醇产率最高,为0.45g/g葡萄糖。他们还研究了废弃棉花热解油加酸水解,实验证明,用0.2mol/L的硫酸在121℃加热反应20min所得葡萄糖产率最高 (17.4%)。稀释后的酸水解液成功地被酿酒酵母发酵,并且所得乙醇产率比纯净葡萄糖发酵所得的乙醇产率要高。Prosen等[86]也在利用木材热解物时发现了同样的现象。他们还研究了一种提高酵母菌对热解生物油中有毒物质的耐受性的预适应策略,通过12次复筛后得到一株高耐受的酵母菌株,它可以用来发酵葡萄糖含量高得多的水解液 (95.8g/L),并且与其亲代菌株相比,乙醇的产率提高了47%。
对于从欧洲赤松热解油的水解液中分离出的LGA,Bennett等[11]用0.5mol/L硫酸在125℃条件下水解44min,得到葡萄糖产率最高,达216%(基于最初的LGA),由此可知,在水解反应中其他葡萄糖衍生物也被转化为了葡萄糖。葡萄糖含量高达20%的水解液成功地被酿酒酵母S. cerevisiae T2发酵,所得的乙醇产率 (0.46g/g葡萄糖) 与理论计算的用纯净的葡萄糖应得产率 (0.51g/g葡萄糖) 十分接近,说明用热解油发酵产乙醇跟用纯净的葡萄糖发酵产乙醇同样高效。
Chan等[63]研究了酿酒酵母S. cerevisiae T2菌株在有氧和无氧条件下利用生物油水解后的水解液发酵生产乙醇。提取LGA的最优方法是用相同质量的水来萃取热解油,得到的LGA产率最高为热解油的4.98(wt)%。采用Bennett等[11]的方法进行水解,水解液用氢氧化钙过度中和后,以40%的浓度用于发酵,得到最高的乙醇产率 (0.45g/g葡萄糖)。有氧条件下,驯化后的酵母菌所得的乙醇产率比未驯化的酵母菌所得的乙醇产率高39%;无氧条件下,驯化后的酵母菌所得的乙醇产率比未驯化的酵母菌所得的乙醇产率高26%。
Lian等[68]提出了一种利用脱水糖生产油脂和乙醇的策略。首先用乙酸乙酯作为溶剂来分离热解糖和酚类物质,然后酸催化水解脱水糖为葡萄糖,水解液用氢氧化钡中和,并用活性炭脱毒以去除发酵抑制物,最终利用弯曲隐球菌Cryptococcus curvatus和红酵母Rhodotorula glutinis发酵产油脂,酿酒酵母S. cerevisiae发酵产乙醇。乙醇产率高达0.473g/g葡萄糖,比油脂产率 (0.167g/g葡萄糖) 要高得多,这说明热解糖用于乙醇发酵比用于脂质发酵更高效。
纵观近来关于微生物利用生物质热解产物生产生物乙醇的相关研究,不难发现生物质热解油的生物技术应用挑战在于高效的脱毒策略,另外,可直接利用LGA的微生物缺乏也限制其发展。因此,研究者致力于开发高效的重组菌株利用LGA作为碳源。
4.2 LGA的直接利用
4.2.2 基因工程菌利用LGA
从自然界中分离出的微生物没有工业微生物那么高效,而且这些天然的微生物总是缺乏可以直接利用LGA的基因和代谢途径,因此需要投入大量的科学研究使它们变为有用的“生物工厂”[92]。目前的研究表明,酿酒酵母S. cerevisiae和大肠杆菌Escherichia coli是基因工程改造和代谢工程改造的首选工业微生物,因为它们的基因组已经被充分确证过并被长期应用于工业生产[92]。4.1节中所述的LGA的间接发酵,发酵前必须用硫酸水解LGA为葡萄糖,这不仅增加成本而且还产生热量,以至于生成附加的发酵抑制物[77]。针对这种情况,有两种可选择的解决办法:一种是从自然界中筛选能直接、高效利用LGA的微生物,但是这相当费时费力;另一种选择则是采用基因工程技术向现有的发酵微生物中导入LGA的代谢途径。目前已经有一些关于向其他微生物中导入LGA直接代谢途径的研究。
Kim等[17]提供了第一个在谷氨酸棒状杆菌 (一种广为人知的工业氨基酸生产菌) 中利用LGA生产琥珀酸的实例。由于野生型谷氨酸棒状杆菌不能利用LGA作为唯一碳源,所以将斯达油脂酵母菌、玉米赤霉菌、铜绿假单胞菌中编码LGK的一条基因在谷氨酸棒状杆菌中异源表达。结果,只有来源于斯达油脂酵母菌的LGK基因在谷氨酸棒状杆菌中表达才能利用LGA作为唯一碳源在CgXII基本培养基中催化LGA转化为葡萄糖-6-磷酸。随后,LGK基因在琥珀酸生产菌株BL-1中表达,重组菌株的琥珀酸产率比参照菌株更高,重组菌株在2%LGA中产率为0.25g/g,参照菌株BL-1在2%葡萄糖中产率为0.19g/g,这确证了LGA对于谷棒生产菌株来说是一种有吸引力的碳源。
酵母菌株L. starkeyi中含有LGK,它对LGA有很高的催化活性,能将其转化为葡萄糖-6-磷酸[93]。Dai等[18]识别、克隆、并在E. coli中表达了一条来自酵母菌L. starkeyi YZ-215的LGK基因的cDNA,得到的工程菌株可以在基本培养基中将LGA作为唯一的碳源。Layton等[74]通过密码子最优化后克隆LGK基因得到了一个产乙醇的E. coli基因工程菌,这个工程菌不仅将LGA作为碳源和能源,还将其转化为乙醇。这些研究都说明,利用基因工程技术,现存的微生物有望轻易地转变为LGA的直接利用者。
为了加强微生物对LGA的直接利用,一方面我们要从环境中分离出具有新的LGA直接代谢途径的微生物,另一方面我们可以进一步进行基因工程修饰改造,以使微生物在少量毒副作用物质存在的条件下能够生长,并且具备直接将LGA转化为乙醇等产物的能力。
4.2.1 真菌利用LGA
除了生物乙醇,LGA也被用来作为碳源发酵生产一些其他有价值的化合物。许多真核微生物和原核微生物可以通过它们自身的LGA代谢途径直接代谢LGA,生产有价值的产物[66, 75, 78, 79]。目前已经识别出了许多可以利用LGA作为唯一碳源和能源的微生物[79]。
研究表明,真核微生物中LGA的代谢途径比原核生物中的要成熟和先进。原核生物 (如节细菌属Arthrobacter sp.) 以NAD+为辅酶因子,至少通过3步酶催化反应代谢LGA (如图式4所示)。与之相反,真核生物 (例如丝状真菌和酵母菌) 则通过LGK将LGA直接转化为葡萄糖-6-磷酸 (如图式3所示)[75, 78, 79]。葡萄糖-6-磷酸发生一系列氧化反应生成丙酮酸,最终在丙酮酸脱氢酶复合体的作用下生成乙酰辅酶A。在无氧条件下,丙酮酸在丙酮酸脱羧酶的作用下生成乙醛,最终以NADH为辅酶因子,在乙醇脱氢酶的作用下乙醛被转化为乙醇。由于LGK的存在,许多真菌和酵母菌能够直接代谢作为唯一碳源和能源的LGA[66, 75, 86]。土曲霉Aspergillus terreus K26可发酵LGA产衣康酸[66],黑曲霉Aspergillus niger CBX-209可以在含有LGA的基质中生长并将LGA转化为柠檬酸[78]。这两个例子中振奋人心的地方在于它们的发酵速率和产物产率与纯净葡萄糖发酵相似,这说明微生物利用LGA跟利用传统的六碳糖 (葡萄糖或果糖) 一样高效。
5 结语
综上所述,LGA在生物技术方面的应用还不够深入。除了看到LGA极具潜力的应用前景,我们更应该关注高效利用LGA所面临的挑战。所以,在今后的研究工作中,一方面要致力于开发环境友好、经济划算、易于工业化、易于回收提取、能够显著提高生物质分解中LGA产率的化学工艺,例如在溶剂体系中分解生物质;另一方面就是要从自然界中筛选或者利用基因工程技术开发更多能高效利用LGA作为唯一碳源和能源的微生物,并且研究确证LGA在微生物体内的代谢机理。总之,LGA作为一种新型糖源,对生物技术、化学工程的发展都是有很大的实际意义的。
-
-
[1]
S Czernik, A Bridgwater. Energy Fuels, 2004, 18(2):590-598. doi: 10.1021/ef034067u
-
[2]
M Chuang, C K Chou, K Sopajaree et al. Atmosph. Environ., 2013, 78:72-81. doi: 10.1016/j.atmosenv.2012.06.056
-
[3]
E Hopmans, R L Santos, A Mets et al. Org. Geochem., 2013, 58:86-88. doi: 10.1016/j.orggeochem.2013.02.003
-
[4]
N Kehrwald, R Zangrando, P Gabrielli et al. Tellus B, 2012, 64:1-9.
-
[5]
Y Tsai, K Sopajaree, A Chotruksa et al. Atmosph. Environ., 2013, 78:93-104. doi: 10.1016/j.atmosenv.2012.09.040
-
[6]
K Vancampenhout, B Vos, K Wouters et al. Soil Biol. Biochem., 2012, 50:40-46. doi: 10.1016/j.soilbio.2012.03.005
-
[7]
P Wallner, M Kundi, H Moshammer et al. Int. J. Hyg. Environ. Health, 2013, 216(3):280-283. doi: 10.1016/j.ijheh.2012.05.001
-
[8]
T B Jordan, A J Seen, G E Jacobsen. Atmosph. Environ., 2006, 40(27):5316-5321. doi: 10.1016/j.atmosenv.2006.03.023
-
[9]
J T Overpeck. Nature, 1992, 356(6371):670-670.
-
[10]
A Vicente, C Alves, A I Calvo et al. Atmosph. Environ., 2013, 71:295-303. doi: 10.1016/j.atmosenv.2013.01.062
-
[11]
N M Bennett, S S Helle, S J B Duff. Bioresour. Technol., 2009, 100(23):6059-6063. doi: 10.1016/j.biortech.2009.06.067
-
[12]
X Hu, L Wu, Y Wang et al. Green Chem., 2012, 14(11):3087-3098. doi: 10.1039/c2gc35961h
-
[13]
X L Zhuang, H X Zhang, J J Tang. Biomass Bioenergy, 2001, 21(1):53-60. doi: 10.1016/S0961-9534(01)00012-5
-
[14]
Y Zhao, Y Chen, H Sun et al. Acta Microbiol. Sin., 2014, 54(7):821-827.
-
[15]
Z Yu, J Ning, L Zhang et al. Chin. Agr. Sci. Bull., 2011, 27(6):342-349.
-
[16]
J R Klesmith, J Bacik, R Michalczyk et al. ACS Synth. Biol., 2015, 4(11):1235-1243. doi: 10.1021/acssynbio.5b00131
-
[17]
E Kim, Y Um, M Bott et al. FEMS Microbiol. Lett., 2015, 362(19):1-6.
-
[18]
J Dai, Z Yu, Y He et al. World J. Microbiol. Biotechnol., 2009, 25(9):1589-1595. doi: 10.1007/s11274-009-0048-9
-
[19]
D Chang, Z Yu, Z Islam et al. Appl. Microbiol. Biotechnol., 2015, 99(9):4093-4105. doi: 10.1007/s00253-015-6475-7
-
[20]
S Kersten, M Perez. Curr. Opin. Biotechnol., 2013, 24(3):414-420. doi: 10.1016/j.copbio.2013.04.003
-
[21]
A V Bridgwater, D Meier, D Radlein. Org. Geochem., 1999, 30(12):1479-1493. doi: 10.1016/S0146-6380(99)00120-5
-
[22]
A V Bridgwater. Biomass Bioenergy, 2012, 38:68-94. doi: 10.1016/j.biombioe.2011.01.048
-
[23]
C A Mullen, A A Boateng. Energy Fuels, 2008, 22(3):2104-2109. doi: 10.1021/ef700776w
-
[24]
T Hosoya, S Sakaki. ChemSusChem, 2013, 6(12):2356-2368. doi: 10.1002/cssc.201300338
-
[25]
X Zhang, J Li, W Yang et al. Energy Fuels, 2011, 25(8):3739-3746. doi: 10.1021/ef2005139
-
[26]
X Zhang, W Yang, W Blasiak. J. Anal. Appl. Pyrolysis, 2012, 96:110-119. doi: 10.1016/j.jaap.2012.03.012
-
[27]
X Zhang, W Yang, W Blasiak. Fuel, 2012, 96(1):383-391.
-
[28]
X Zhang, W Yang, W Blasiak. Fuel, 2013, 109:476-483. doi: 10.1016/j.fuel.2013.03.035
-
[29]
X Zhang, W Yang, C Dong. J. Anal. Appl. Pyrolysis, 2013, 104:19-27. doi: 10.1016/j.jaap.2013.09.015
-
[30]
X Bai, P Johnston, S Sadula et al. J. Anal. Appl. Pyrolysis, 2013, 99:58-65. doi: 10.1016/j.jaap.2012.10.028
-
[31]
A Zheng, Z Zhao, Z Huang et al. Green Chem., 2015, 17(2):1167-1175. doi: 10.1039/C4GC01724B
-
[32]
L K Tolonen, M Juvonen, K Niemela et al. Carbohydr. Res., 2015, 401:16-23. doi: 10.1016/j.carres.2014.10.012
-
[33]
J Piskorz, D S Radlein, D S Scott et al. J. Anal. Appl. Pyrolysis, 1989, 16(2):127-142. doi: 10.1016/0165-2370(89)85012-0
-
[34]
B Pecha, P Arauzo, M Garcia-Perez. J. Anal. Appl. Pyrol., 2015, 114:127-137. doi: 10.1016/j.jaap.2015.05.014
-
[35]
S R G Oudenhoven, R J M Westerhof, N Aldenkamp et al. J. Anal. Appl. Pyrol., 2013, 103:112-118. doi: 10.1016/j.jaap.2012.10.002
-
[36]
E Le Roux, M Chaouch, P N Diouf et al. Biomass Bioenergy, 2015, 81:202-209. doi: 10.1016/j.biombioe.2015.07.005
-
[37]
S Kumagai, R Matsuno, G Grause et al. Bioresour. Technol., 2015, 178:76-82. doi: 10.1016/j.biortech.2014.09.146
-
[38]
L Jiang, A Zheng, Z Zhao et al. Bioresour. Technol., 2015, 182:364-367. doi: 10.1016/j.biortech.2015.01.032
-
[39]
H Xia, X Yan, S Xu et al. J. Chem., 2015, 2015:1-11.
-
[40]
G Kwon, S Kuga, K Hori et al. J. Wood Sci., 2006, 52(5):461-465. doi: 10.1007/s10086-005-0784-x
-
[41]
G Kwon, D Kim, S Kimura et al. J. Anal. Appl. Pyrol., 2007, 80(1):1-5. doi: 10.1016/j.jaap.2006.12.012
-
[42]
Z Yang, X Liu, Z Yang et al. J. Anal. Appl. Pyrol., 2013, 102:83-88. doi: 10.1016/j.jaap.2013.03.012
-
[43]
P R Patwardhan, J A Satrio, R C Brown et al. J. Anal. Appl. Pyrol., 2009, 86(2):323-330. doi: 10.1016/j.jaap.2009.08.007
-
[44]
X Bai, P Johnston, R C Brown. J. Anal. Appl. Pyrol., 2013, 99:130-136. doi: 10.1016/j.jaap.2012.10.012
-
[45]
Q Li, P H Steele, F Yu et al. J. Anal. Appl. Pyrol., 2013, 100:33-40. doi: 10.1016/j.jaap.2012.11.013
-
[46]
A Pictet, J Sarasin. HeIv. Chim. Acta, 1918, 1(1):87-96. doi: 10.1002/hlca.19180010109
-
[47]
F Shafizadeh, T T Stevenson. J. Appl. Polym. Sci., 1982, 27(12):4577-4585. doi: 10.1002/app.1982.070271205
-
[48]
M J Antal, J G Varhegyi. Ind. Eng. Chem. Res., 1995, 34(3):703-717. doi: 10.1021/ie00042a001
-
[49]
C Blasi. Chem. Eng. Sci., 2000, 55(24):5999-6013. doi: 10.1016/S0009-2509(00)00406-1
-
[50]
J P Diebold. Biomass Bioenergy, 1994, 7(1):75-85.
-
[51]
J Lédé. J. Anal. Appl. Pyrol., 2012, 94:17-32. doi: 10.1016/j.jaap.2011.12.019
-
[52]
I Milosavljevic, E M Suuberg. Ind. Eng. Chem. Res., 1995, 34(4):1081-1091. doi: 10.1021/ie00043a009
-
[53]
A R Teixeira, K G Mooney, J S Kruger et al. Energy Environ. Sci., 2011, 4(10):4306-4321. doi: 10.1039/c1ee01876k
-
[54]
N Kuzhiyil, D Dalluge, X Bai et al. ChemSusChem, 2012, 5(11):2228-2236. doi: 10.1002/cssc.201200341
-
[55]
F Cao, T J Schwartz, D J Mcclelland et al. Energy Environ. Sci., 2015, 8(6):1808-1815. doi: 10.1039/C5EE00353A
-
[56]
M Ohara, A Takagaki, S Nishimura et al. Appl. Catal. A-Gen., 2010, 383(1-2):149-155. doi: 10.1016/j.apcata.2010.05.040
-
[57]
C R Vitasari, G W Meindersma, A B Haan. Bioresour. Technol., 2011, 102(14):7204-7210. doi: 10.1016/j.biortech.2011.04.079
-
[58]
A V Bridgwater, G V C Peacocke. Renew. Sust. Energ. Rev., 2000, 4(1):1-73. doi: 10.1016/S1364-0321(99)00007-6
-
[59]
A Oasmaa, Y Solantausta, V Arpiainen et al. Energy Fuels, 2010, 24:1380-1388. doi: 10.1021/ef901107f
-
[60]
F Zeng, W Liu, H Jiang et al. Bioresour. Technol., 2011, 102(2):1982-1987. doi: 10.1016/j.biortech.2010.09.024
-
[61]
R H Venderbosch, W Prins. Biofuel. Bioprod. Bior., 2010, 4(2):178-208. doi: 10.1002/bbb.205
-
[62]
D Mohan, C U Pittman, P H Steele. Energy Fuels, 2006, 20(3):848-889. doi: 10.1021/ef0502397
-
[63]
J K S Chan, S J B Duff. Bioresour. Technol., 2010, 101(10):3755-3759. doi: 10.1016/j.biortech.2009.12.054
-
[64]
H Wang, D Livingston, R Srinivasan et al. Appl. Biochem. Biotechnol., 2012, 168(6):1568-1583. doi: 10.1007/s12010-012-9879-1
-
[65]
Y Zhang, T R Brown, G Hu et al. Bioresour. Technol., 2013, 127:358-365. doi: 10.1016/j.biortech.2012.09.070
-
[66]
M Nakagawa, Y Sakai, T Yasui. J. Ferment. Technol., 1984, 62(2):201-203.
-
[67]
Z S Yu, H X Zhang. Bioresour. Technol., 2003, 90(1):95-100. doi: 10.1016/S0960-8524(03)00093-2
-
[68]
J Lian, S Chen, S Zhou et al. Bioresour. Technol., 2010, 101(24):9688-9699. doi: 10.1016/j.biortech.2010.07.071
-
[69]
D Santhanaraj, M R Rover, D E Resasco et al. ChemSusChem, 2014, 7(11):3132-3137. doi: 10.1002/cssc.201402431
-
[70]
Z S Yu, H X Zhang. Bioresour. Technol., 2004, 93(2):199-204. doi: 10.1016/j.biortech.2003.09.016
-
[71]
Z S Yu, H X Zhang. Biomass Bioenergy, 2003, 24(3):257-262. doi: 10.1016/S0961-9534(02)00147-2
-
[72]
J P Bacik, J R Klesmith, T A Whitehead et al. J. Biol. Chem., 2015, 290(44):26638-26648. doi: 10.1074/jbc.M115.674614
-
[73]
Y Kitamura, T Yasui. Agr. Biol. Chem., 1991, 55(2):523-529.
-
[74]
D S Layton, A Ajjarapu, D W Choi et al. Bioresour. Technol., 2011, 102(17):8318-8322. doi: 10.1016/j.biortech.2011.06.011
-
[75]
K Nakahara, Y Kitamura, Y Yamagishi et al. Biosci. Biotechnol. Biochem., 1994, 58(12):2193-2196. doi: 10.1271/bbb.58.2193
-
[76]
Z Ul Islam, Z Yu, E B Hassan et al. J. Ind. Microbiol. Biotechnol., 2015, 42(12):1557-1579. doi: 10.1007/s10295-015-1687-5
-
[77]
S Helle, N M Bennett, K Lau et al. Carbohydr. Res., 2007, 342(16):2365-2370. doi: 10.1016/j.carres.2007.07.016
-
[78]
X L Zhuang, H X Zhang. Protein Expres. Purif., 2002, 26(1):71-81. doi: 10.1016/S1046-5928(02)00501-6
-
[79]
Y Kitamura, Y Abe, T Yasui. Agr. Biol. Chem., 1991, 55(2):515-521.
-
[80]
H Xie, X Zhuang, Z Bai et al. World J. Microbiol. Biotechnol., 2006, 22(9):887-892. doi: 10.1007/s11274-006-9133-5
-
[81]
P Phitsuwan, K Sakka, K Ratanakhanokchai. Biomass Bioenergy, 2013, 58:390-405. doi: 10.1016/j.biombioe.2013.08.027
-
[82]
L R Jarboe, Z Wen, D Choi et al. Appl. Microbiol. Biotechnol., 2011, 91(6):1519-1523. doi: 10.1007/s00253-011-3495-9
-
[83]
P Galletti, F Moretti, C Samori et al. Green Chem., 2007, 9(9):987-991. doi: 10.1039/b702031g
-
[84]
M Kaldstrom, N Kumar, T Heikkila et al. ChemCatChem, 2010, 2(7):717-717. doi: 10.1002/cctc.201090028
-
[85]
K Hattori, T Yoshida, H Nakashima et al. Carbohydr. Polym., 1998, 312(1-2):1-8.
-
[86]
E M Prosen, D Radlein, J Piskorz et al. Biotechnol. Bioeng., 1993, 42(4):538-541. doi: 10.1002/bit.260420419
-
[87]
A E Farrell, R J Plevin, B T Turner et al. Science, 2006, 311(5760):506-508. doi: 10.1126/science.1121416
-
[88]
E Waltz. Nat. Biotechnol., 2008, 26(1):8-9. doi: 10.1038/nbt0108-8
-
[89]
Z Chi, M Rover, E Jun et al. Bioresour. Technol., 2013, 150:220-227. doi: 10.1016/j.biortech.2013.09.138
-
[90]
L Luque, R Westerhof, G Van Rossum et al. Bioresour. Technol., 2014, 161:20-28. doi: 10.1016/j.biortech.2014.03.009
-
[91]
B Sukhbaatar, Q Li, C Wan et al. Bioresour. Technol., 2014, 161:379-384. doi: 10.1016/j.biortech.2014.03.051
-
[92]
H Alper, G Stephanopoulos. Nat. Rev. Microbiol., 2009, 7(10):715-723. doi: 10.1038/nrmicro2186
-
[93]
J Ning, Z Yu, H Xie et al. World J. Microbiol. Biotechnol., 2008, 24(1):15-22. doi: 10.1007/s11274-007-9432-5
-
[1]
-


表 1 由生物质热解制备左旋葡聚糖的部分文献报道
Table 1. Part of references about the preparation of levoglucosan by cellulose pyrolysis
原料 反应器类型 预处理及催化 LGA产率 参考文献 玉米芯 CDS 5200型微型热解反应器 在甘油中微波预处理 (150W,1min) 38% [31] 纤维素 固定床反应器 固体酸催化 22.1% [39] 微晶纤维素 小型管式反应器 380℃、25MPa条件下,超临界水预处理0.4s 低聚糖产率42%,其中LGA浓度为8mg/mL [32] 黄杉木 CDS 5000型微型热解反应器 稀硝酸酸洗预处理后加入0.05%硫酸作催化剂 没有定量 [34] 杨木 流化床反应器 90℃下用5%硫酸酸洗,之后水洗至pH为6.6 30.42% [33] 松木 20℃单级冷凝器 去矿物质化 15.8% [35] 杨木 实验室规模螺旋钻反应器 高压热水预处理 10.56% [36] 木材 石英管反应器 3mol/L硫酸预处理 8.5% [37] 甘蔗渣 微型裂解炉 稀酸预处理 40.5% [38] 微晶纤维素 快速冷却、连续给料裂解炉,减压热解 (5kPa) 70.1%(分析值) [41] 纤维素 真空条件下 (低于1mmHg),特制玻璃热解反应器 50%~55% [40] 纤维素 微型热解反应器 58.78% [43] 纤维素 CDS 5200型微型热解反应器 56% [44] 松木 反应器改造:在冷凝前的蒸汽流中注入水喷雾 稀酸预处理 热解油的16.43% [45] 玉米淀粉 管式炉热解反应器,减压热解 (600Pa) 56%(分析值) [42]  下载: 导出CSV
下载: 导出CSV
表 2 左旋葡聚糖的间接利用 (乙醇发酵) 的部分相关文献
Table 2. Part of references about the indirect use of levoglucosan (alcoholic fermentation)
提高乙醇产率的策略 发酵微生物 最高乙醇浓度/(g/L) 乙醇产量/(g/g葡萄糖) 参考文献 H2SO4水解+Ca (OH)2中和+硅藻土震荡 S.cerevisiae CCTCC 2.399 16.1 0.45 [71] Pichia sp. YZ-1 15.1 0.42 [71] H2O萃取+H2SO4水解+NaOH中和+高接种浓度+微需氧条件 S.cerevisiae T2 未提及 0.46 [11] H2O萃取+H2SO4水解+Ca (OH)2过度中和+有机溶剂萃取+自适应进化 S.cerevisiae T2 未提及 0.45 [63] 生物柴油或乙酸乙酯萃取+H2SO4水解+Ba (OH)2中和+活性炭 S.cerevisiae ATCC 200062 32~35 0.47 [68]
下载: 导出CSV
-








 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 0
- HTML全文浏览量: 0
 下载:
下载:
