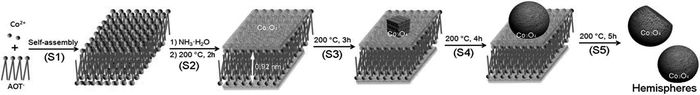
 图1
Co3O4纳米球逐步分裂机制[7]
Figure1.
Co-branching mechanism of Co3O4nanospheres[7]
图1
Co3O4纳米球逐步分裂机制[7]
Figure1.
Co-branching mechanism of Co3O4nanospheres[7]
引用本文:
赵佳伟, 郑志林, 何小伟, 耿旺昌. 不同维度Co3O4纳米材料的水热合成研究进展[J]. 化学通报,
2017, 80(12): 1093-1103.

Citation: Zhao Jiawei, Zheng Zhilin, He Xiaowei, Geng Wangchang. Research Progress in Hydrothermal Synthesis of Different Dimensions Co3O4 Nanomaterials[J]. Chemistry, 2017, 80(12): 1093-1103.
Citation: Zhao Jiawei, Zheng Zhilin, He Xiaowei, Geng Wangchang. Research Progress in Hydrothermal Synthesis of Different Dimensions Co3O4 Nanomaterials[J]. Chemistry, 2017, 80(12): 1093-1103.
不同维度Co3O4纳米材料的水热合成研究进展
English
Research Progress in Hydrothermal Synthesis of Different Dimensions Co3O4 Nanomaterials
Abstract:
Crystal structure and surface characteristics of transition metal oxide Co3O4 are closely related to its morphology, size and microstructure. In this paper, different morphology Co3O4 nanomaterial could be divided into several dimensions through scale, including 0D of nanospheres, cubes, polyhedral, 1D of nanowires, nanorods, nanotubes, nanocolumns and others, 2D of nanometer film, nano flakes and 3D of microscale superstructure constituted by nano-element such as urchin, micron ball etc. We discussed the influencing factors in formation of morphology, focused on the hydrothermal synthesis process and summarized the possible mechanism, to facilitate the deeply understanding of preparation of different dimension materials. Accordingly, it would provide a guidance for the synthesis of materials with specific dimension and morphology.
-
Key words:
- Dimension
- / Co3O4 morphology
- / Solvothermal
- / Growth mechanism
-
四氧化三钴(Co3O4)具有典型的尖晶石结构,是一种稳定且储量丰富的过渡金属氧化物,近年来其纳米材料在低温催化CO氧化[1]、锂离子电池电极材料[2]、超级电容器[3]、磁性材料[4]、电解水催化剂[5]、气敏材料[6]等众多领域有着广泛的应用。Co3O4的应用与本身的电子特性、磁性、光学性能、催化性能密切相关,而这些取决于材料的晶体结构和表面特性。表面特性被形貌、尺寸和微观结构控制,目前已制备出了纳米球[7]、立方块[8]、纳米片[9]、纳米棒[10]、分层结构[11]等形貌结构的Co3O4纳米材料。
可以通过化学气相沉积[12]、脉冲激光沉积[13]、微波辅助[14]、喷雾热解[15]、电沉积法[16]等方法制备出各种不同形貌的Co3O4纳米材料。然而这些制备方法存在诸如高煅烧温度、长反应时间、低产率、步骤繁琐及仪器设备昂贵等缺点,阻碍了其在工业上的应用与发展。与这些方法比较,具有低温、易于控制、大规模合成等特点的液相法,兼具重现性好、适合研究形貌性能关系的优点,成为一种常用和有效的方法。不过,水热法存在的问题在于影响因素太多,如反应物种类与添加量、添加剂的种类与添加量、反应时间、反应温度等因素都会影响产物的最终形貌。为了得到尺寸较为均一、形貌规则的产物,需要对其形成过程乃至形成机理做深入的研究和探索。本文从维度上对溶剂法制备出来的纳米材料进行分类总结。
1 零维(0D)纳米材料
0D纳米材料主要包括球形颗粒和各种纳米多面体,由低指数晶面包围而成。由于垂直于高指数晶面方向的生长速度远大于沿低指数晶面的,进而在颗粒形成过程中高指数晶面快速消失。
1.1 纳米球
纳米球的形成机理较为明确,在无封盖剂或是非离子表面活性剂的作用下合成。Zhao等[17]以葡萄糖为模板、硫酸钴为钴源,水热条件下制备了直径约为200nm的Co3O4空心球,测得其比表面积为49.1m2/g。他们提出了纳米球形成的机理:水溶性葡萄糖溶液在180℃下进行芳构化反应和碳化过程并遵循LaMer模型[18],得到了带有亲水性基团和活性表面的碳球;随后加入硫酸钴和作为新的碳源的葡萄糖,碳球亲水基团和活性表面进一步发生脱水、碳化过程,在这个过程中,钴离子被嵌入新形成的碳层;经过过滤和煅烧,得到了Co3O4空心球。
对于纳米球形貌形成的过程存在有不同的解释。Gong等[7]以琥珀酸2-乙基己基酯磺酸钠(AOT)为表面活性剂制备了平均直径为250nm的多孔单晶Co3O4纳米球。他们通过研究反应条件,提出了一种逐步分裂机制来解释Co3O4纳米球的形成(图 1):首先,AOT与Co2+形成由AOT双层和两个Co2+层状组装单元组成的纳米反应器(S1);钴离子在水热条件下经过2h水解形成Co3O4纳米晶(S2);延长反应时间,在Ostwald熟化作用下纳米片转化为由长方体组成的树枝状纳米立方体(S3);纳米立方体逐步分裂生长最终得到纳米球(S4、S5)。AOT在纳米球形成中的作用是,(1)形成层状组装作为纳米反应器;(2)限制纳米片的生长;(3)指导长方体选择性连接在纳米片表面形成纳米球。
He等[19]用组分相同的三份溶液分别在搅拌、回流、超声后水热条件下制备了不同尺寸的Co3O4纳米空心球。他们提出了中间产物分解的机理来解释Co3O4空心球的形成:在水热条件下首先形成了球形的中间产物碱式硝酸钴(CHN),这种球形的CHN颗粒是由50nm纳米片状CHN定向聚集而成;随着反应的进行,定向聚集的CHN纳米球转变为Co3O4空心球。
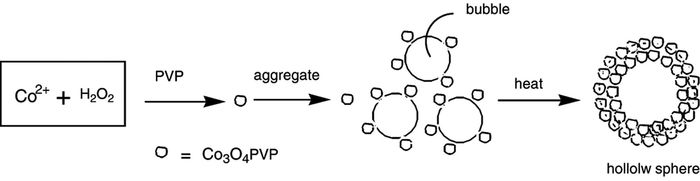
Chen等[20]在聚乙烯吡咯烷酮(PVP)存在下制备了平均直径为350nm、壳厚约为40nm的Co3O4空心球。他们探索了表面活性剂对材料形貌的影响,以等量的十二烷基苯磺酸钠(SDBS)、十六烷基三甲基溴化铵(CTAB)替代PVP,分别得到了球形及大块团聚的颗粒,不添加任何表面活性剂则得到了球形颗粒。他们认为球形颗粒的形成归因于原始Co3O4纳米晶的定向团聚。H2O2和水中的PVP形成氢键被吸附,同时钴离子与PVP形成不稳定的PVP-Co2+复合物,两者在PVP上发生氧化还原反应形成Co3O4纳米晶。在最低化界面能量的推动下,纳米晶沿着这个过程产生的CO2气泡和溶液之间的气固相界面团聚形成空心球(图 2),球的大小取决于气泡的尺寸。
纳米球的组成可影响材料的性能。Liu等[21]以铜离子作为结构导向剂,在不添加任何表面活性剂情况下制备了平均直径为400nm的单分散多孔Co3O4纳米球。SEM照片显示纳米球是由厚度为几个纳米的纳米片组成。为了进一步证明铜离子的作用,以氯化铜、硫酸铜取代醋酸铜得到的仍是球形形貌。相比于商业Co3O4粉末,将其作为超级电容器的电极材料显示出更加优异的性能。
1.2 八面体
八面体形貌是通过对暴露晶面的生长调控而得到的。Zhou等[22]采用三苯基膦(PPh3)的辅助水热法制备出了长约为70nm、宽约为60nm的Co3O4八面体。依据对反应条件的探索,Co3O4八面体的形成可以分成三个步骤:首先,NaOH加入后Co(OH)2絮状物的形成;水热条件下Co(OH)2絮状物溶解和缩聚形成Co3O4小颗粒;包含有PPh3的生长单元吸附在Co3O4晶核的(111)晶面上,减缓该方向上的生长速率,经过重结晶和Ostwald熟化过程形成Co3O4八面体。对于其机理分析借鉴了Li等[23]提出的晶体生长模型和晶体界面模型。初始阶段钴离子主要以[Co(OH)4]2-形式存在,部分被氧气氧化为[Co(OH)6]3-。上述离子中的羟基基团的脱水缩合从而形成了Co3O4晶核,[Co(OH)x]h-x分散并吸附在其表面,羟基之间相互反应形成新的表面。依据界面模型,大的离域π键的PPh3相比水分子更易与钴离子形成配合物[Co(OH)a(H2O)b(PPh3)x-a-b]h-a,随着反应时间延长,含有PPh3的生长单元更多被吸附在表面。由于(111)晶面具有最大的表面羟基密度,[Co(OH)a(H2O)b(PPh3)x-a-b]h-a将会被优先吸附,并以PPh3取代其表面的羟基形成大的空间位阻,进而限制其生长速度,形成了附着(111)晶面的八面体。
2012年,Liu等[24]首次通过聚合物辅助水热法合成了凹面Co3O4八面体介晶纳米材料。与一般八面体光滑的表面不同,具有明显界面纳米颗粒的粗糙表面表明其由原始的20~50 nm的纳米颗粒组装而成。由于纳米晶体的形貌由(111)晶面方向生长速度和(001)晶面方向的生长速度之比来决定,比值为1.73时得到八面体。他们[25]认为取向粘附生长的介晶形貌形成同样取决于两个晶面方向生长速率之比。不同于纳米晶体是由单个纳米颗粒的粗化而成,介晶生长是由原始纳米颗粒的定向聚合而成。聚(4-苯乙烯磺酸钠)(PSS)键接在(001)晶面上作为Co3O4生长单元的基石,生长单元上的苯环存在相互作用导致了原始纳米晶体在同样的6个(100)方向上的取向粘附,形成了Co3O4八面体。而(111)晶面只有一个方向被PSS包覆,粘附在这个方向上阻碍较强,使得(111)晶面方向生长速度和(001)晶面的生长速度的比值低于1.73,从而导致了凹面八面体的形成。
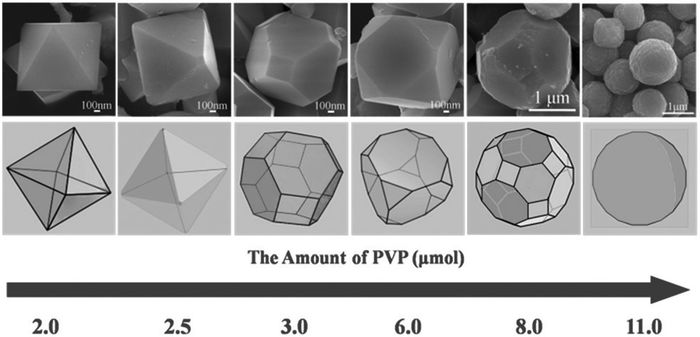
一些特殊配体可以与钴配位,从而强烈影响Co3O4纳米粒子的生长。Kang等4[26]利用表面活性剂辅助法合成了一系列Co3O4纳米晶体,通过改变表面活性剂的量制备了包括八面体、角截面八面体、角截面立方体、球状多面体和微球等特殊形貌,在实验的基础上,他们提出了Co3O4晶核的部分特殊面通过选择性吸附C2O42-和PVP引起晶体界面限制效应使得形貌演化的解释(图 3)。其中,随着PVP用量的增加,Co3O4由八面体向微球转变。
1.3 纳米立方块
纳米立方块由于具有占主要的(001)暴露晶面立方结构,便于研究晶面控制的相关性能如催化剂的活性和选择性而受到重视。低单体浓度控制的低速率生长是纳米立方块形成的一个重要因素,热力学因素在形貌控制上起了重要作用。
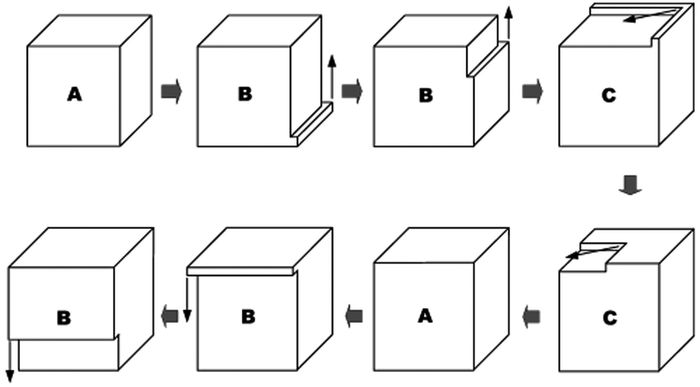
Zeng等[27]以硝酸钴为钴源,以水为溶剂并添加硝酸钠在95℃下水热法制备了约为47nm的Co3O4立方块,尺寸较为均一。与不添加硝酸盐的序列对比,添加后形成了具有刻面的立方块。因为较高浓度的硝酸钠可以大幅降低氧气在体系中的溶解度,同时溶液中的离子强度增加形成了盐-溶剂表面的扩散边界,阻碍钴的氧化过程和界面生长。Zeng等[28]以同样的方法,通过改变水热温度和时间得到了边长10~100 nm的立方块,建立了立方块尺寸和反应时间的线性关系。如表 1,随着反应时间的增加,立方块的边长显著增大;而水热温度的升高可以明显加快立方块的形成过程。在实验基础上,他们提出了一种“表面包裹”的理论来描述立方块形貌控制生长过程(图 4):整个生长流程为…→A smaller→B→C→A larger→…,在这个立方块生长过程中整个结构的各向同性严格地保持不变。立方块在长大过程中要经历B和C两个阶段,如同A被一层层壳包裹。
Aging/h Mean size/nm Standard dev./nm Percent dev./% 90g of sodium nitrate used 3 11.2 1.6 14.3 6 24.3 2.1 8.7 12 44.7 2.8 6.3 150g of sodium nitrate used 3 10.6 1.4 13.2 6 25.3 1.7 6.7 12 43.1 3.7 8.6 He等[29]以Co(NO3)2·4H2O为钴源,采用液相氧化还原过程制备了2nm球状和2.5、4.7 nm立方块状的Co3O4单分散纳米晶。他们提出了溶解度控制过程来解释超细纳米晶体的形成,并得出在溶解度控制的过程中立方块纳米晶的尺寸和反应时间成正比。晶体的成核和生长过程可由紫外可见光检测跟踪,发现通过调节硝酸钠的溶解度可以分隔开这两个阶段,并由此制备了立方块状的颗粒。基于单体浓度随反应时间的变化,胶体颗粒的形成可以分为三个阶段:单体浓度增加、成核和生长阶段。形成单分散颗粒的关键在于成核和生长阶段的分离。当反应速度控制在一个适当值时,成核后的单体浓度难以达到二次成核的过饱和度。另一方面,在溶解和氧化还原同时进行过程中,Co3O4单体持续提供并且始终处于成核和颗粒继续生长的浓度之间,直至硝酸钠被完全消耗。这导致颗粒间的Ostwald熟化将不能发生,并形成了尺寸均一的颗粒。反应时间延长后,随着硝酸钠被消耗完,颗粒间的粗化将会发生,平均尺寸大约为10nm且粒度分布显著加大。大部分的立方块形貌变得不规整,主要是因为边缘和顶端的原子因为高的化学势优先溶解。
Lou等[30]以硝酸钴为钴源、PVP为表面活性剂,水热条件下制备了尺寸约为350nm的表面光滑的立方块。他们认为在非离子型表面活性剂PVP的参与下,立方块形貌形成遵循“定向附着”机理。这个过程中在现有的水热条件下形成尺寸较小的纳米立方块,表面活性剂吸附在其表面阻止其不规则的团聚。延长反应时间,尺寸小的立方块将会与邻近未被PVP完全包覆的立方块结合,形成较大的立方块。
近年,通过对晶面的控制来进行形貌调控受到了关注。Xiao等[31]通过一步水热法合成了三种不同形貌(立方体、截断八面体、八面体)的Co3O4。此过程不添加任何的封端剂,简单改变Co(NO3)2·6H2O和NaOH的量就可以很好地控制Co3O4形貌。依据面心纳米晶体的形貌由(111)晶面方向生长速度和(001)晶面的生长速度之比来决定,他们对这一过程进行了解释。当反应物量较少时可能会促进晶面沿着(111)晶面生长从而形成带有(001)晶面的立方体;控制NaOH量不变,增加Co(NO3)2·6H2O,(111)晶面生长速度减缓而(001)晶面生长速度增加,产物形貌便会由立方体向具有(001)和(111)的截断面八面体转变;两者的量都进一步增加时,得到暴露(111)晶面的八面体。总之,高浓度的NaOH将会有利于(111)的稳定性(图 5)。进一步研究显示,八面体的电化学性能优于截断八面体,截断八面体又优于立方体。
晶面对材料物化性能起较大作用。Hu等[32]在以硝酸钴为钴源、乙醇为溶剂,添加十八烯胺的水热条件下制备了尺寸约为15~20 nm的均一单分散纳米立方块。通过改变反应条件则又制备出了六边形片和2~5 μm长、50~100 nm宽的纳米带。立方块、纳米带、片的主要暴露晶面分别为(001)、(011)、(112),比表面积分别为22.6、20.1、17.8 m2·g-1。在313℃下甲烷催化燃烧实验中三者的转化率分别为23%、42%、50%。得出了这些晶面中甲烷催化燃烧的催化活性顺序为(112)>(011)≫(001)。他们的实验表明,在一定条件下,晶面对性能的影响要大于比表面积的影响。
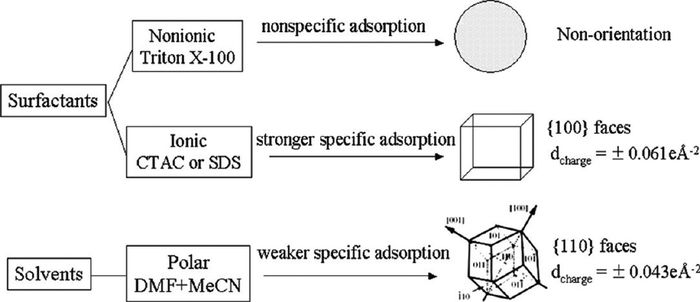
不同表面活性剂对多面体的形成也有一定影响。Sasaki等[33]采用不同带电表面活性剂在溶剂水热条件下制备了400nm的纳米球、约150nm的纳米立方块及平均直径约为200nm的斜方十二面体(图 6)。当非离子型TritonX-100加入时,中性链无选择性地吸附在晶体表面上,形成了无特定晶面取向Co3O4球。他们认为十六烷基三甲基氯化铵(CTAC)或十二烷基硫酸钠(SDS)与(100)的强相互作用导致了纳米立方块的形成。在离子型表面活性剂CTAC或SDS的存在下,带电R4N+和SO2-优先吸附在高带电(100)晶面,阻碍其在该晶面上的生长从而形成Co3O4纳米立方块。DMF和丙酮与CTAC和SDS相比是中性或弱的覆盖剂,其与(100)相互作用较弱,将不能弥补因阻碍(100)晶面生长所引起系统能量的升高,从而吸附在(110)晶面上形成了斜方十二面体。该机理分析能够适用于之前所报道纳米立方块的合成,这些合成过程会存在部分高带电的覆盖剂,如SDBS、来源于Tween-85的阴离子或是高浓度的NaNO3。
总之,对于0D Co3O4纳米材料而言,无特殊取向的纳米球是在无封盖剂或是非离子表面活性剂的作用下合成的;立方块、八面体等多面体则是表面活性剂和封盖剂吸附在某一暴露晶面进而控制不同晶面的生长速度而得到的。
2 一维(1D)纳米结构
2.1 纳米线
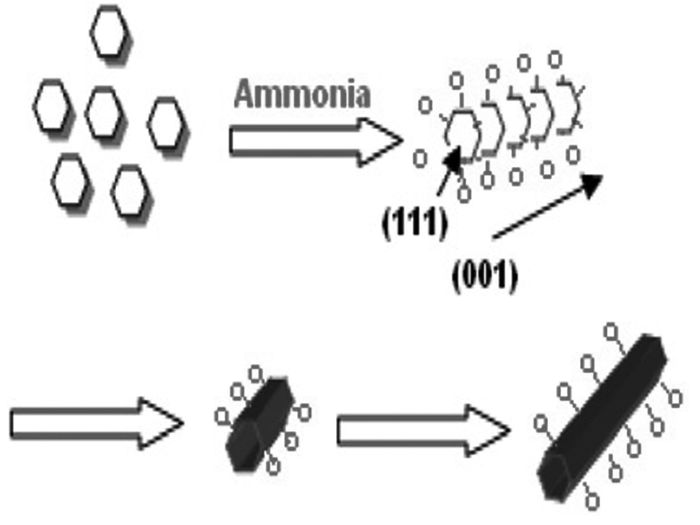
纳米线是典型的1D形貌。Waleed等[34]以钴粉和H2O2为反应物,在氨水的调节下水热制备了直径5nm、长度10μm的超细β-Co(OH)2纳米线,煅烧后得到Co3O4纳米线。氨水在β-Co(OH)2纳米线的形成中起着重要作用,它改变了Ehrlich-Schwoebel势垒和吸附原子的迁移率,使生长沿着最低能量的(001)面进行(图 7)。
纳米线的生长方向受溶液离子的影响。Wang等[35]通过水热法制备了碱式碳酸钴前驱体纳米线,在煅烧下制备了多孔Co3O4纳米线。通过改变尿素的浓度来调控前驱体的形貌和结构。尿素缓慢分解产生的OH-和CO32-可能导致纳米线沿着长轴生长。尿素浓度较低时得到分散性较好的纳米线,升高浓度得到的纳米线粘连成束状,浓度继续升高将会得到直径为5μm的纳米线向四周辐射的海胆状微米球。他们推测产生的CO32-对纳米线的各向异性生长起到重要作用,并能够作为复杂分层结构的粘连剂。
纳米线可由基本单元组装而成。Xia等[36]以包覆有5nm厚的Co3O4镍箔、硅片、镍泡沫为基底,进行水热实验大面积制备了自支撑的空心纳米线阵列。在氧气氛围下,纳米片紧密堆积在一起形成了一维纳米柱,然后堆叠在一起的单个纳米片整合在一起形成纳米线。空心纳米线的形成是轴螺旋位错和Kirkendall效应的综合结果。产生于晶体表面的轴螺旋位错效应为粘附表面吸附的原子提供活性位点,加速沿着螺旋位错方向晶体的生长从而形成了纳米线状结构。Kirkendall效应导致缩聚形成空隙,空隙在外离子快速交换的平衡下向内流动,形成空心结构。
2.2 纳米棒、纳米带、纳米管和纳米片
2.2.1 纳米棒
纳米棒具有较低的纵横比。Nguyen等[37]在不添加任何表面活性剂或模板剂情况下,仅通过CoCl2和尿素的水热法制备了具有介孔/大孔的Co3O4纳米棒。Co3O4沿着不同取向生长和粘连形成了多孔纳米棒,直径约为200nm,长度达到几个微米,孔尺寸主要集中在2.18、36.9、284 nm。对所得纳米棒进行挥发性气体气敏测试,其显示出对丙酮较好的敏感性、快的响应恢复性能。
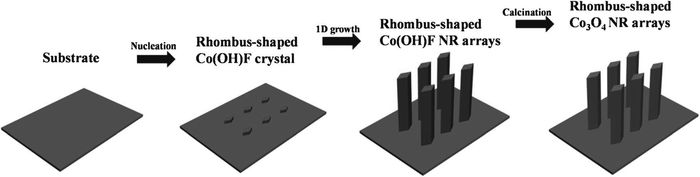
Zhu等[38]通过简单氟介导水热法在镍泡沫基底上制备了边长约为400nm的菱形Co(OH)F纳米棒阵列前驱体,在煅烧下转化为多孔Co3O4纳米棒阵列。其表面粗糙且有孔,孔的形成可能是热处理过程中脱水和晶格收缩造成的。其形成过程如图 8所示,首先,钴离子和氟离子形成复合物;水热温度达到95℃时,HMT水解提供OH-,与复合物形成Co(OH)F晶体;镍泡沫悬挂功能基团作为锚点在氢键作用下捕获Co(OH)F,Co(OH)F晶体团聚形成菱形纳米棒。NH4F的加入能阻止Co(OH)2的形成,Co(OH)2具有水矿石层状结构容易形成纳米片。他们以陶瓷气敏原件为基底用同一方法制备了尺寸相近的纳米棒[39],其对乙醇的敏感度达到了70.7。高气敏性归因于纳米棒的独特结构:尺寸可调的孔使气体更易扩散并吸附在其表面;大比表面积提供更多的活性位点供气体吸附和转移。
2.2.2 纳米带
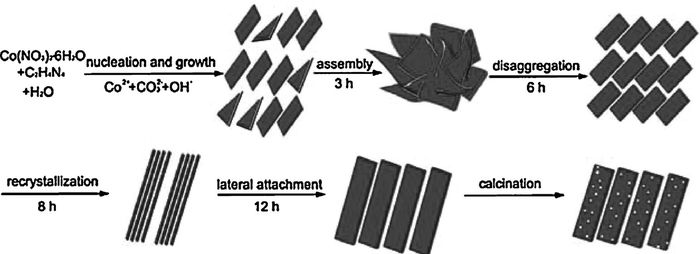
Che等[40]用无模板的水热法制备出前驱体,随后对其进行热处理得到了1D多孔Co3O4纳米带。结果发现,实验参数如Co(NO3)2·6H2O与C2H4N4的摩尔比、Co(NO3)2·6H2O的浓度、水热温度对产物的形貌和结构有着较大影响。摩尔比为0.5或1时,得到了片状结构;摩尔比为3时得到带状结构;摩尔比增加到4时得到了无规则的片状和块状混合物。他们提出了一种可能的形成过程,如图 9所示。随着反应时间的增加,形貌经历了由花状微观结构到线状纳米结构的转变。第一个阶段,Co2+和CO32-在OH-存在下形成前驱体核,核在高表面能的推动下团聚生长成为片状结构。纳米片组装成3D微米花,随后发生重结晶形成2D碎片,进一步转化为1D纳米线,由于纳米线的侧向粘附而形成纳米带。最后经过高温煅烧,释放CO2、H2O后形成多孔纳米带。与商业Co3O4粉末相比,多孔Co3O4纳米带显示出对乙醇更好的气敏性能。
Huang等[41]利用水热法制备了宽100~300 nm、长为几个微米的Co3O4纳米带,该纳米带具有独特的由20~30 nm连通纳米晶组成的介孔结构。通过水热过程形成前驱体并热转化为Co3O4。混合前驱体为Co(OH)F和碱式碳酸钴,其可能形成过程为:水热条件下,钴离子与氟离子形成CoF+和CoFx(x-2)-复合物;水热温度上升到120℃尿素水解,缓慢产生了CO32-和OH-;OH-与CoF+形成Co(OH)F,CO32-、OH-与CoFx(x-2)-形成碱式碳酸钴。该纳米带作为锂电池材料显示出了高的存储容量和快的充电能力,特别是具有优异的循环稳定性。层状1D纳米结构确保了电子的快速传递,并为锂离子的嵌入/脱出提供了较短的路径;多孔结构则能增加电解质与电极的接触面积,并为嵌入/脱出过程提供大的体积变化,进而提高了循环性能。
2.2.3 纳米管
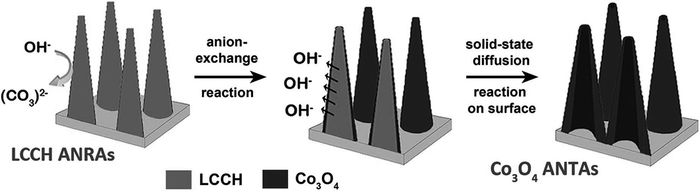
与1D Co3O4纳米棒和纳米线阵列相比,1D纳米管具有相似的电子传递优势,还因为具有内表面提供了更大的表面积。Kung等[42]低温下在导电石墨基底上两步无模板法合成了1D Co3O4针状纳米管阵列(ANTAs)。首先在基底上通过化学浴沉淀法于90℃下制备了层状钴酸氢氧化物(LCCH)的针状纳米棒阵列(ANRAs),然后在95℃下NaOH水热处理制备了Co3O4 ANTAs。将LCCH在350℃下煅烧,得到Co3O4针状纳米棒阵列(ANRAs)。Co3O4 ANTAs和ANRAs都是由多晶组成的,顶部尺寸为50nm,底部为300nm。由于氢氧化钴和碳酸钴的溶解度不同,加入NaOH后LCCH自发与OH-反应,同时发生离子交换。在该水热条件下,Co3O4比氢氧化钴更加稳定,离子交换的结果使得在纳米棒的表面形成Co3O4薄层。包在Co3O4壳里面的LCCH为了继续与加入的OH-反应而发生固相分散。核心LCCH的浓度梯度越大,提供的向外扩散的推动力越大,依据Kirkendall效应,不同的扩散速率会导致Kirkendall空隙的形成。LCCH不断向Co3O4壳外扩散,与OH-反应,导致Co3O4壳加厚,最终形成管状结构(见图 10)。
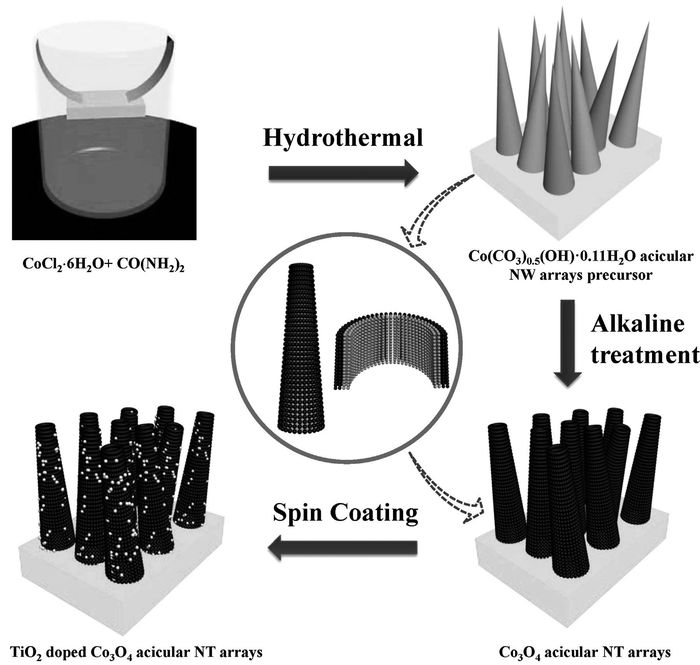
将不同种类的纳米材料复合是提高传感器性能的一个有效途径。Gao等[43]通过水热法合成得到了Co3O4 ANTAs,之后用TiO2纳米颗粒涂覆形成新的葡萄糖传感器(图 11)。由于Co3O4 ANTAs内表面具有的大表面积以及TiO2纳米颗粒提供的反应位点,显示出在碱性溶液中对葡萄糖检测的显着性能。所以对Co3O4纳米材料进行掺杂改性是提升其性能的较为直接的途径。
2.2.4 纳米片
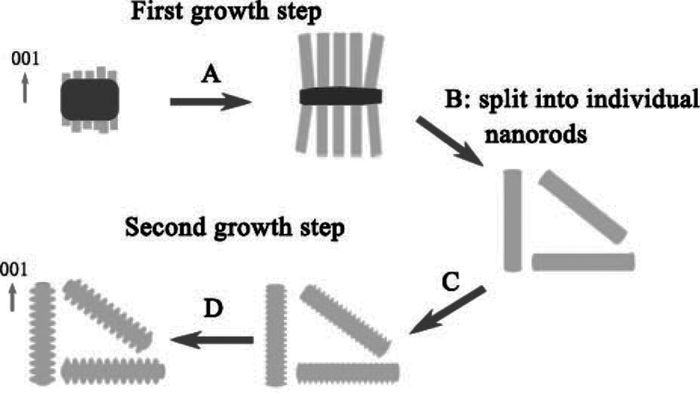
Shao等[44]用水合肼和Na3PO4作为形貌导向剂,通过一步水热法制备了高产量的一维分层β-Co(OH)2纳米柱,经过煅烧得到Co3O4纳米柱。磷酸盐离子更易吸附在垂直于(001)面的晶面上,作为抑制剂减缓纳米晶的生长速度,导致晶体沿着(001)晶面生长。水合肼使得棒状物上出现很多片状突起,为纳米晶的生长提供高能位点,钴离子自发地在这些位点上成核,形成纳米片。β-Co(OH)2纳米柱的形成分为两步,纳米棒的形成及薄的纳米片横向生长。图 12为β-Co(OH)2纳米柱在400℃退火4h下获得的Co3O4形态。
纳米柱的形成机理见图 13。(A)第一步生长可以看做成核-溶解-重结晶过程。随着反应温度的升高,亚稳态的块状物快速形成,归因于内部固有的分层结构和与水合肼的配合。同时棒状的Co(OH)2突起出现在块状物表面。(B)块状物在碱性环境中逐渐溶解并提供钴离子形成纳米棒。Na3PO4的存在是重结晶形成纳米棒的关键,不加Na3PO4只能得到纳米片。随着时间的延长,纳米棒变长,最终完全溶解形成了单个纳米棒。(C)在水合肼和气液平衡的作用下出现片状突起,为纳米晶的生长提供很多高能位点,钴自发地的在这些位点上成核形成纳米片。(D)水合肼在分层的形成中具有结构导向的作用[44, 45],因为其本身为分层结构和双齿配体(在同等环境下用氨水替换水合肼只能得到纳米片)。最终纳米片形成纳米柱,在Ostwald熟化作用下形成均一和完整的结构。
对于1D Co3O4纳米材料而言,大多数都是沿低能量晶面生长得到的,部分物质(NH3·H2O、OH-、CO32-等)能够显著影响晶面的能量,从而对生长方式进行调控。相对于0D材料,1D纳米材料具有较好的电子传递能力,尤其是纳米管具有大内表面积,在传感、电容等领域有着十分广阔的应用前景。
3 二维(2D)纳米结构
Wang等[46]通过氨辅助的水热法,经高温煅烧制备了雪花状Co3O4纳米片(Co3O4-SF),该纳米片宽10μm、厚100nm,纵横比达到了100。氨水是合成的重要因素,通过改变其浓度可使材料的形貌转变为六边形纳米片(Co3O4-HX),边长为500nm,厚约为90nm。当氨水与钴离子的摩尔比为3.4:1时,水热条件下得到六边形纳米片。氨水是前驱体Co(OH)2形成的沉淀剂和结构控制剂。NH4+能够控制Co(OH)2沿着主要暴露晶面(001)生长。当氨水与钴离子的摩尔比达到10:1时,形貌转变为雪花状,此时氨水除了作为前驱体Co(OH)2形成的沉淀剂和结构导向剂外,还是雪花状纳米片形成的配体。作为锂离子电池材料,Co3O4-SF优异于Co3O4-HX和其他结构,原因在于小的纳米颗粒和超薄的结构能够减少分散时间,且暴露的活性面有利于缩短分散长度以及较大的纵横比能够维持高效的电子传输路径。
高维度形貌有利于部分电学性能。Lee等[47]通过PVP控制的水热法制备了表面带孔的2D纳米片,被用作可充电的锌-空气电池材料。其形成是因为PVP作为封盖剂吸附在特定晶面上阻碍其生长,使得晶体生长被限制在两个维度上。2D多孔纳米片作为电极材料显示出较为优异的充放电电压及循环稳定性,这源于其特殊形貌有利于电解过程中的传质扩散和电荷转移。
Wang等[48]以醋酸钴为钴源,水热条件下合成了Co2CO3(OH)2超薄纳米片,煅烧后得到了厚度为20~50 nm的Co3O4纳米片。首先,尿素在水解过程中同时提供OH-和CO32-,沉淀过程形成Co2CO3(OH)2晶核,晶核生长并团聚成纳米片。第二个阶段,Co2CO3(OH)2纳米片在高温下分解释放出CO2和H2O气体,形成多孔Co3O4超薄纳米片。该纳米片比表面积高达97.2m2/g,为气体和材料之间提供了更多的活性反应位点,进而在对气体敏感性测试中响应快且具有高的灵敏度。
Yan等[49]利用溶剂热法将钴离子与吡嗪和1, 3, 5-均苯三羧酸(BTC)配位形成金属有机框架(Co-CPs)前驱体,经过煅烧得到了由纳米结构团聚而成的Co3O4 2D纳米结构。其形貌由众多的水热条件控制(图 14)。水热较短时间得到了完好的纳米片状(Co-CP-A),之后纳米片自组装成打开书本状的(Co-CP-B),继续反应得到了重叠的纳米片(Co-CP-C)。其中打开书本状的纳米Co3O4在多次循环高放电速率下显示出较高的电容量。较好的电性能在于该形貌能够为锂离子的分散提供高效的途径,并增加了电解液与Co3O4接触界面的面积。
 图14
(a) Co-CP-A、(b)Co-CP-B和(c)Co-CP-C的SEM图像,(d)Co-CP随着反应时间的演变和多孔Co3O4聚集体示意图; (a′)Co3O4-A,(b′)Co3O4-B和(c′)Co3O4-C的SEM图像[49]
Figure14.
SEM images of (a) Co-CP-A, (b) Co-CP-B and (c) Co-CP-C. (d) Schematic illustration showing the evolution of the Co-CPs with reaction time and the formation of the porous Co3O4aggregates. SEM images of (a′) Co3O4-A, (b′) Co3O4-B and (c′)Co3O4-C[49]
图14
(a) Co-CP-A、(b)Co-CP-B和(c)Co-CP-C的SEM图像,(d)Co-CP随着反应时间的演变和多孔Co3O4聚集体示意图; (a′)Co3O4-A,(b′)Co3O4-B和(c′)Co3O4-C的SEM图像[49]
Figure14.
SEM images of (a) Co-CP-A, (b) Co-CP-B and (c) Co-CP-C. (d) Schematic illustration showing the evolution of the Co-CPs with reaction time and the formation of the porous Co3O4aggregates. SEM images of (a′) Co3O4-A, (b′) Co3O4-B and (c′)Co3O4-C[49]
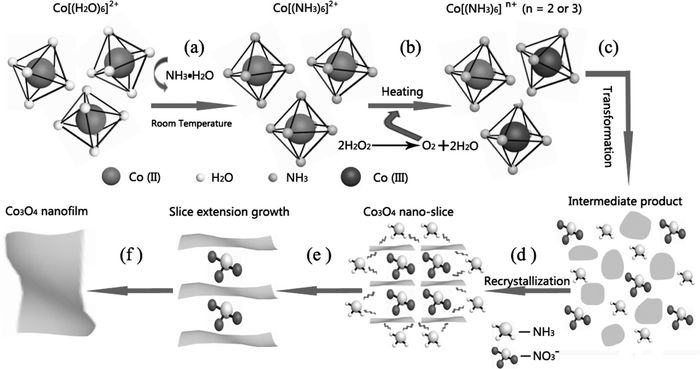
Feng等[50]采用无基底、无表面活性剂的水热法大量制备了小于3nm厚的Co3O4纳米薄膜。超薄纳米膜的形成突出显现了钴-氨复合物的重构及氨和硝酸根对于薄膜生长控制的综合作用(图 15)。首先,依据配位理论,钴离子吸附六个配体形成具有八面体结构的金属复合物。(a)在室温过程中,形成了Co[NH3]62+;(b)由于螯合稳定系数Co[NH3]63+远大于Co[NH3]62+,在水热条件下部分Co[NH3]62+分解,而由于H2O2分解另外部分会被氧化为更加稳定的Co[NH3]63+;(c)Co[NH3]6n+在沸水中,发生重构形成无数的的纳米薄层;重结晶过程得到Co3O4纳米片;水热过程中硝酸根插入钴-氧键之间作为抑制剂减缓其沿着(100)面方向的生长速率,在这种条件下生成的Co3O4纳米片沿着2D方向融合,最终形成原子层Co3O4纳米薄膜。该材料在初次充放电过程中显示出高达1400F/g的比电容,并在1500次循环测试过后仍保持了稳定的电容量。
2D Co3O4纳米材料的形成过程与1D类似,均为在不同溶剂以及添加剂条件下的晶体生长过程。由于2D的片状结构通常能为反应提供较大的接触面积,提供了更多的活性位点,提高了分散程度,在电容、电池材料方面受到研究者的广泛关注。
4 三维(3D)纳米结构
合成低维度纳米晶体后利用这些纳米结构单元来组装成分层结构,也即3D超结构。这种原位自组装的方法被认为是合成超结构的自下而上的有效途径。
Liu等[51]采用无表面活性剂辅助水热法合成了各种形貌的分层碱式碳酸钴前驱体,随后煅烧得到了3D分层介孔Co3O4。通过调节溶剂中乙二醇/水的摩尔比可以在一定程度上控制所得前驱体的形貌,提高乙二醇比例可使构建3D材料的基本单元由针状转变为片状。Wang等[52]利用水热法制备了3D花状α-Co(OH)2微米结构前驱体,经过煅烧得到了由六边形多孔纳米片组装而成的3D花状分层结构。在90℃水热条件下六亚甲基四胺分解为甲醛和氨,钴离子与氨水解形成OH-的反应生成Co(OH)2晶核。在NaCl的作用下,晶核生长为2D片状结构,并螺旋组装成3D花状分层结构。NaCl的具体作用需要进一步探讨。Yang等[53]通过两步溶剂热法制备了海胆状和束状的3D分层介孔Co3O4材料,并通过控制时间和反应温度探索其形成过程。他们认为前驱体的形貌转变经过了定向粘附机理和Ostwald熟化过程。首先碱式碳酸钴核彼此相连经过自发的自组装形成了1D纳米片。在定向粘附机理作用下,形成了2D纳米片,最终形成了3D海胆结构。合成的海胆状和束状材料应用于锂离子电池显示出优异的电化学性能,3D结构和介孔的存在为锂离子和电子提供有效的分散途径,并缓冲锂离子的嵌入/脱出造成的体积膨胀。Kong等[54]通过原位反应合成了泡沫Ni支持下的独特三维Co3O4纳米片结构,发现其在高速率下具有较大的比容量和优异的电化学稳定性,该方法将为开发电池型电容器材料提供指导。
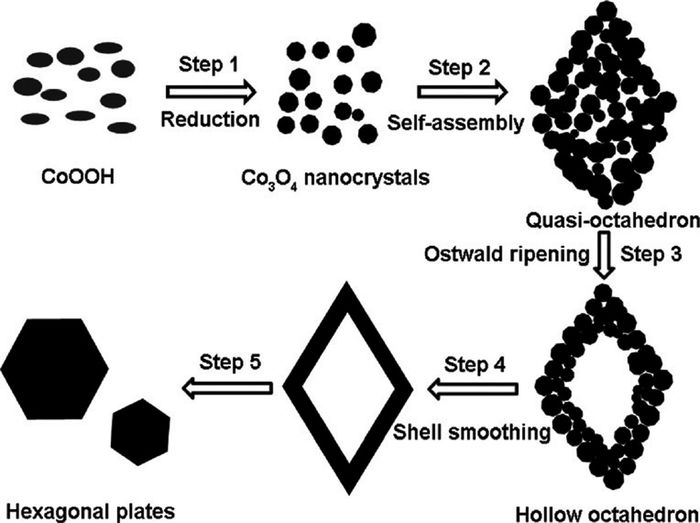
Cao等[55]采用一步水热法制备了空心八面体Co3O4,其通过原始纳米晶的自组装形成八面体结构,后通过Ostwald熟化作用形成空心结构。通过分析提出其结构和几何形貌的一个演化过程(图 16):(1)NaOH加入首先与钴离子形成Co(OH)2-,搅拌过程中被水溶氧氧化为深褐色的CoOOH,其在水热下逐渐被转化为Co3O4纳米颗粒;(2)在无表面活性剂体系中晶体的形貌取决于具有最低表面能的表面; 对于面心立方晶来说(111)晶面的表面能最小,因而纳米颗粒倾向于自组装成具有六个(111)面的八面体;(3)在Ostwald熟化下准八面体的核心将会溶解,并在外表面重结晶以降低表面能;(4)组成八面体的原始纳米晶融合在一起,八面体表面变得光滑,表明八面体壳由介晶转化为了单晶。原始纳米晶融合形成光滑表面在于纳米颗粒表面向纳米颗粒间的颈区的传质过程。
原位自组装的方法被认为是合成3D超结构的自下而上的有效途径。三维Co3O4纳米材料的形成过程可以理解为2D材料的组装,通常是在2D的基础上在结构导向剂的作用下完成,由于结构的复杂性,该类材料的应用范围较广,不同形貌的3D结构Co3O4纳米材料研究也是当今的研究热点。
5 结语
本文概述了现阶段不同维度Co3O4纳米材料的溶剂热合成方法、生长机理及其性能应用。在多维度Co3O4材料研究方面已取得了较大进展,主要通过控制其暴露的晶面(各个晶面生长速度不同)合成了0D(纳米球、立方块、多面体等)、1D(纳米线、纳米棒、纳米管、纳米柱等)、2D(纳米片、纳米薄膜等)、3D(海胆状、微米球等)材料。但是研究维度与性能的关系,还未见详细报道。此外,笔者认为以下两方面有待深入研究:(1)精确的合成条件才能制备出形貌均一、特定晶面暴露的纳米Co3O4材料,其生长机理和理论研究需要进一步探明;(2)结合理论与实践,如采用Vienna Ab-initio Simulation Package、Material studio等模拟软件进行晶面控制预测,进而为溶剂热合成提供指导。
-
-
[1]
X Xie, Y Li, Z Q Liu et al. Nature, 2009, 458:746~749. doi: 10.1038/nature07877
-
[2]
P Poizot, S Laruelle, S Grugeon et al. Nature, 2000, 407:496~499. doi: 10.1038/35035045
-
[3]
L Bao, T Li, S Chen et al. Small, 2017, 13(5):1602077. doi: 10.1002/smll.v13.5
-
[4]
N Rinaldi-Montes, J González-López, Á Fernández-González et al. Ceram. Int., 2017, 43:10889~10894. doi: 10.1016/j.ceramint.2017.05.125
-
[5]
H Hu, B Guan, B Xia et al. J. Am. Chem. Soc., 2015, 137:5590~5595. doi: 10.1021/jacs.5b02465
-
[6]
D Zhang, C Jiang, P Li et al. ACS Appl. Mater. Interf., 2017, 9:6462~6471. doi: 10.1021/acsami.6b15669
-
[7]
Y Gong, F Gong, C Wang et al. RSC Adv., 2015, 5:27266~27272. doi: 10.1039/C5RA02739J
-
[8]
R Xu, H C Zeng. Langmuir, 2004, 20:9780~9790. doi: 10.1021/la049164+
-
[9]
D Su, S Dou, G Wang. Nano Res., 2014, 7:794~803. doi: 10.1007/s12274-014-0440-0
-
[10]
A C Nwanya, D Obi, R U Osuji et al. J. Solid State Electrochem., 2017, 21(9):2567~2576. doi: 10.1007/s10008-017-3520-8
-
[11]
D T Dam, J M Lee. ACS Appl. Mater. Interf., 2014, 6:20729~20737. doi: 10.1021/am506660q
-
[12]
N Bahlawane, P H T Ngamou, V Vannier et al. Phys. Chem. Chem. Phys., 2009, 11:9224~9232. doi: 10.1039/b910707j
-
[13]
R Edla, S Gupta, N Patel et al. Appl. Catal. A, 2016, 515:1~9. doi: 10.1016/j.apcata.2016.01.031
-
[14]
W Yao, Q Dai, Y Liu et al. ChemElectroChem, 2017, 4(1):230~235. doi: 10.1002/celc.201600564
-
[15]
R Manogowri, R M Mathelane, S Valanarasu et al. J. Mater. Sci.:Mater. Electron., 2016, 27:3860~3866. doi: 10.1007/s10854-015-4234-2
-
[16]
W H Ryu, T H Yoon, S H Song et al. Nano Lett., 2013, 13:4190~4197. doi: 10.1021/nl401868q
-
[17]
W Zhao, Y Liu, H Li et al. Mater. Lett., 2008, 62:772~774. doi: 10.1016/j.matlet.2007.06.057
-
[18]
G C Luijkx, F van Rantwijk, H van Bekkum et al. Carbohyd. Res., 1995, 272:191~202. doi: 10.1016/0008-6215(95)00098-E
-
[19]
T He, D Chen, X Jiao et al. Langmuir, 2004, 20:8404~8408. doi: 10.1021/la0488710
-
[20]
Y Chen, Y Zhang, S Fu. Mater. Lett., 2007, 61:701~705. doi: 10.1016/j.matlet.2006.05.046
-
[21]
R Liu, Z Jiang, Q Liu et al. CrystEngComm, 2015, 17:4449~4454. doi: 10.1039/C5CE00658A
-
[22]
H Zhou, B Lv, D Wu et al. CrystEngComm, 2013, 15:8337~8344. doi: 10.1039/c3ce41419a
-
[23]
W Li, E Shi, W Zhong et al. J. Synth. Cryst., 1999, 28:117~125.
-
[24]
Y Liu, G Zhu, B Ge et al. CrystEngComm, 2012, 14:6264~6270. doi: 10.1039/c2ce25788b
-
[25]
S E Skrabalak, Y Xia. ACS Nano, 2009, 3:10~15. doi: 10.1021/nn800875p
-
[26]
M Kang, H Zhou, D Wu et al. CrystEngComm, 2016, 18:9299~9306. doi: 10.1039/C6CE02106A
-
[27]
R Xu, H C Zeng. J. Phys. Chem. B, 2003, 107:926~930. doi: 10.1021/jp021094x
-
[28]
J Feng, H C Zeng. Chem. Mater., 2003, 15:2829~2835. doi: 10.1021/cm020940d
-
[29]
T He, D Chen, X Jiao et al. Chem. Mater., 2005, 17:4023~4030. doi: 10.1021/cm050727s
-
[30]
J S Chen, T Zhu, Q H Hu et al. ACS Appl. Mater. Interf., 2010, 2:3628~3635. doi: 10.1021/am100787w
-
[31]
X Xiao, X Liu, H Zhao et al. Adv. Mater., 2012, 24:5762~5766. doi: 10.1002/adma.v24.42
-
[32]
L Hu, Q Peng, Y Li. J. Am. Chem. Soc., 2008, 130:16136~16137. doi: 10.1021/ja806400e
-
[33]
J Yang, T Sasaki. Cryst. Growth Design, 2010, 10:1233~1236. doi: 10.1021/cg9012284
-
[34]
W E Mahmoud, F Al-Agel. J. Phys. Chem. Solids, 2011, 72:904~907. doi: 10.1016/j.jpcs.2011.04.014
-
[35]
B Wang, T Zhu, H B Wu et al. Nanoscale, 2012, 4:2145~2149. doi: 10.1039/c2nr11897a
-
[36]
X Xia, J Tu, Y Mai et al. J. Mater. Chem., 2011, 21:9319~9325. doi: 10.1039/c1jm10946d
-
[37]
H Nguyen, S A El-Safty. J. Phys. Chem. C, 2011, 115:8466~8474. doi: 10.1021/jp1116189
-
[38]
W Mei, J Huang, L Zhu et al. J. Mater. Chem., 2012, 22:9315~9321. doi: 10.1039/c2jm00123c
-
[39]
Z Wen, L Zhu, W Mei et al. Sens. Actuat. B, 2013, 186:172~179. doi: 10.1016/j.snb.2013.05.093
-
[40]
H Che, A Liu, J Hou et al. Mater. Res. Bull., 2014, 59:69~76. doi: 10.1016/j.materresbull.2014.06.033
-
[41]
H Huang, W Zhu, X Tao et al. ACS Appl. Mater. Interf., 2012, 4:5974~5980. doi: 10.1021/am301641y
-
[42]
C W Kung, Y H Cheng, C M Tseng et al. J. Mater. Chem. A, 2015, 3:4042~4048. doi: 10.1039/C4TA06811D
-
[43]
Z Gao, L Zhang, C Ma et al. Biosens. Bioelectron., 2016, 80:511~518. doi: 10.1016/j.bios.2016.02.004
-
[44]
Y Shao, J Sun, L Gao. J. Phys. Chem. C, 2009, 113:6566~6572. doi: 10.1021/jp9005718
-
[45]
C Coudun, J F Hochepied. J. Phys. Chem. B, 2005, 109:6069~6074. doi: 10.1021/jp0466441
-
[46]
B Wang, X Y Lu, Y Tang. J. Mater. Chem. A, 2015, 3:9689~9699. doi: 10.1039/C5TA00140D
-
[47]
D U Lee, J Scott, H W Park et al. Electrochem. Commun., 2014, 43:109~112. doi: 10.1016/j.elecom.2014.03.020
-
[48]
X Wang, S Yao, X Wu et al. RSC Adv., 2015, 5:17938~17944. doi: 10.1039/C4RA14450C
-
[49]
B Yan, L Chen, Y Liu et al. CrystEngComm, 2014, 16:10227~10234. doi: 10.1039/C4CE01277A
-
[50]
C Feng, J Zhang, Y He et al. ACS Nano, 2015, 9:1730~1739. doi: 10.1021/nn506548d
-
[51]
P Liu, Q Hao, X Xia et al. J. Phys. Chem. C, 2015, 119:8537~8546.
-
[52]
W Wang, J Xu. ACS Appl. Mater. Interf., 2014, 7:415~421.
-
[53]
T Yang, Y Liu, Z Huang et al. RSC Adv., 2015, 5:24486~24493. doi: 10.1039/C4RA16871B
-
[54]
S Kong, F Yang, K Cheng et al. J. Electroanal. Chem., 2017, 785:103~108. doi: 10.1016/j.jelechem.2016.12.002
-
[55]
Y Cao, F Yuan, M Yao et al. CrystEngComm, 2014, 16:826~833. doi: 10.1039/C3CE41840E
-
[1]
-








图 14 (a) Co-CP-A、(b)Co-CP-B和(c)Co-CP-C的SEM图像,(d)Co-CP随着反应时间的演变和多孔Co3O4聚集体示意图; (a′)Co3O4-A,(b′)Co3O4-B和(c′)Co3O4-C的SEM图像[49]
Figure 14 SEM images of (a) Co-CP-A, (b) Co-CP-B and (c) Co-CP-C. (d) Schematic illustration showing the evolution of the Co-CPs with reaction time and the formation of the porous Co3O4aggregates. SEM images of (a′) Co3O4-A, (b′) Co3O4-B and (c′)Co3O4-C[49]

-




 下载:
下载:















 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 0
- HTML全文浏览量: 0
 下载:
下载:
