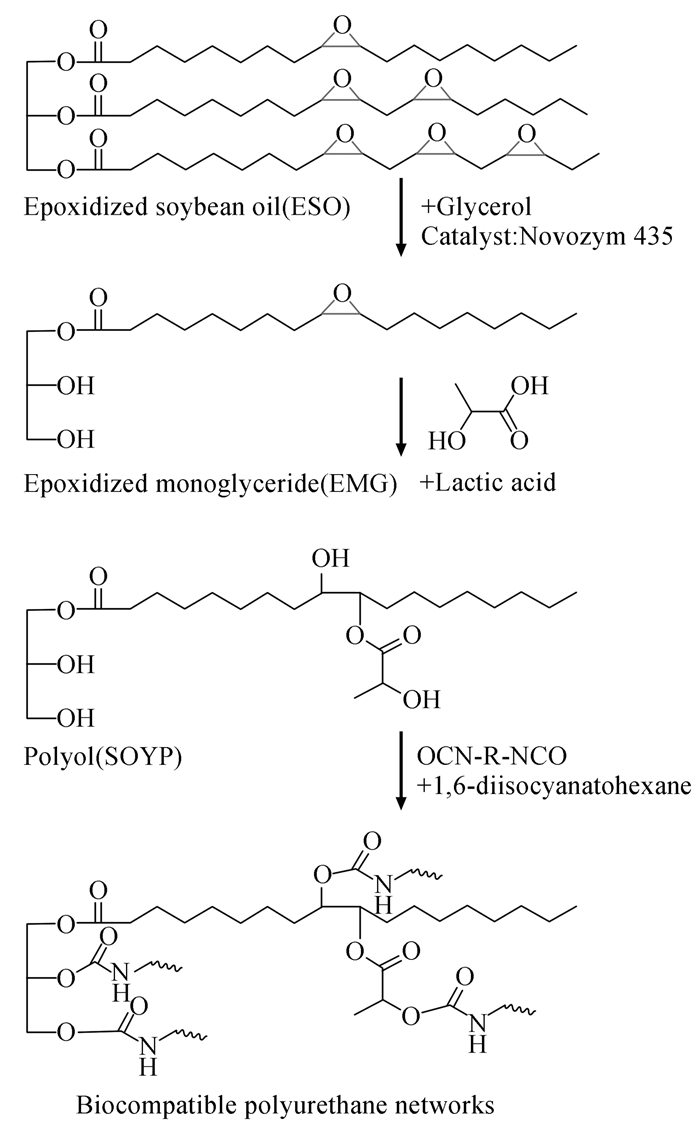
 图式 1
生物基聚氨酯网络的合成路线[13]
Scheme1.
Reaction scheme for the synthesis of biocompatible PU neteorks[13]
图式 1
生物基聚氨酯网络的合成路线[13]
Scheme1.
Reaction scheme for the synthesis of biocompatible PU neteorks[13]
引用本文:
张军瑞, 高煜, 蒋国军. 功能聚氨酯的研究进展[J]. 化学通报,
2017, 80(11): 1027-1035.

Citation: Zhang Junrui, Gao Yu, Jiang Guojun. Research Progress in Functional Polyurethanes[J]. Chemistry, 2017, 80(11): 1027-1035.
Citation: Zhang Junrui, Gao Yu, Jiang Guojun. Research Progress in Functional Polyurethanes[J]. Chemistry, 2017, 80(11): 1027-1035.
功能聚氨酯的研究进展
English
Research Progress in Functional Polyurethanes
Abstract:
Polyurethanes are a class of special polymers with high tunable properties, and have been widely used in many fields due to the versatility in selection of raw materials. In this article, the trends in the development status and research progress of high performance polyurethanes were introduced, the application and research orientation for functional polyurethanes were summarized, and the application problems of polyurethanes in different fields were discussed. Finally, the development prospect of functional polyurethane materials in different fields was prospected.
-
Key words:
- Polyurethane
- / Biomedical
- / Electricity
- / Shape memory
- / Mechanical response
-
聚氨酯材料是由德国化学家Bayer在20世纪30年代发明的,1940年聚氨酯主要以弹性体和泡沫的产品类型进入市场,并迅速在市场中占有重要的地位。随后,聚氨酯产品以其原材料多样、配方灵活、产品形式多样、性能优异且可自主调控等优点,在各行各业中的应用越来越广泛[1~3]。传统的聚氨酯主要以聚氨酯泡沫塑料、结构弹性体、涂料、胶粘剂、革树脂和助剂形式存在。随着科研的进步,聚氨酯材料开始向新的方向发展,例如:采用植物类原材料淀粉、植物油、天然橡胶、糖浆、纤维素、葡萄糖等合成可生物降解的聚氨酯[4];聚氨酯独特的物理性能、水解稳定性、低体外蛋白吸附和血小板粘附等性能使其在生物医学领域得到了广泛的应用[5];此外,导电聚氨酯、形状记忆聚氨酯、机械响应聚氨酯、抗菌防污等领域也是新型聚氨酯的研究热点。本文总结了生物基聚氨酯、导电聚氨酯、形状记忆聚氨酯、机械响应聚氨酯的最新研究与动态,并展望了功能聚氨酯未来的发展方向。
1 生物基聚氨酯
生物基聚氨酯材料具有环境友好、容易获得、低成本及可生物降解等优点,近年来得到广泛的研究和应用。其合成所需要的两类主要原材料多元醇和异氰酸酯可以采用植物油、腰果壳油、桉树焦油等生物可再生资源制备,而自然界丰富的物质资源可以合成出各种各样结构的多元醇和异氰酸酯,赋予生物基聚氨酯结构多变、性能各异的特点[6]。
1.1 植物油基聚氨酯
植物油价格低廉、容易获得、可再生且可转基因生产,其中最常见的是大豆油、蓖麻油、菜籽油、葵花籽油、玉米油等。蓖麻油自身含有羟基官能团,可以直接与异氰酸酯反应制备聚氨酯,但其羟基活性小且官能度低,导致所制备聚氨酯材料的机械性能较差[7]。使用植物油制备聚氨酯主要通过以下两种途径:(1)以植物油为原材料合成聚氨酯用多元醇,以环氧-开环制备多元醇的方法最常用,因其在结构中引入了伯羟基或仲羟基,使植物油基多元醇的羟值大幅度提升,从而使反应活性增强且聚氨酯的交联密度增加[8];(2)以植物油为原材料合成聚氨酯用异氰酸酯。传统异氰酸酯采用光气法制备,对人体和环境具有较大的危害,以植物油为原材料可开发出环境友好型异氰酸酯[9~11]。植物油基异氰酸酯制备的聚氨酯因异氰酸酯中有较长的烷基链常具有较好的拉伸强度[12]。通过对植物油合成的多元醇和异氰酸酯进行合理的结构设计,可以制备出具有更好塑性、刚度和形状的聚氨酯材料。
Miao等[13, 14]采用环氧化的单甘油酯和乳酸以环氧-开环方法合成聚氨酯用多元醇,所制备的聚氨酯(合成路线见图式 1)具有较好的机械性能、生物相容性和耐酸碱等优异性能。Hojabri等[10]采用大豆油为原料,以两种合成工艺制备了两种结构的大豆油基异氰酸酯并在实验室成功用于聚氨酯材料的制备,但其合成路线较长且成本较高。除此之外,腰果壳油、桉树焦油等也可以合成具有特殊优异性能的聚氨酯材料[15~17]。
1.2 多糖基聚氨酯
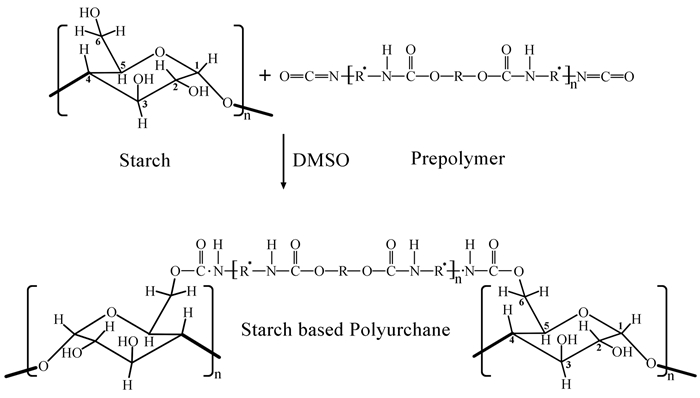
1.2.1 淀粉基聚氨酯
淀粉是一类天然、可再生、来源丰富且低成本的生物降解材料,它可来源于很多植物,如玉米、木薯、小麦、谷物、水稻和马铃薯等。根据来源植物的不同,淀粉颗粒的大小、组成、形态和超分子排列方式可能会有所不同。淀粉分为直链淀粉和支链淀粉两大类,直链淀粉含有几百个葡萄糖单元,支链淀粉含有几千个葡萄糖单元。在天然淀粉中,直链的占20%~26%,它是可溶性的,其余的则为支链淀粉。淀粉的分子链中含有大量羟基,可与异氰酸酯反应制备聚氨酯(图式 2),淀粉的独特性质使淀粉基聚氨酯在生物医学材料领域具有良好的应用前景。淀粉基聚氨酯常用的制备方法包括共混、接枝、共聚和纳米复合等[18]。
Yang等[19]采用氧化的玉米淀粉制备了水性聚氨酯,并对其机械性能及疏水性能进行了研究,结果表明,玉米淀粉使聚氨酯的结构和形态发生显著变化,进而对聚氨酯的机械性能产生明显的影响。当玉米淀粉的加入量为10(wt)%,可使聚氨酯的拉伸强度提高到14.9MPa,吸水率降至4.9%。Lee等[20]使用以乙烯基三甲氧基硅烷改性的淀粉成功制备了可生物降解的聚氨酯,这种方法的主要缺点是淀粉和其他原材料存在相容性问题。Cao等[21]以蓖麻油基聚氨酯和20(wt)%的苯甲基直链淀粉合成了半互穿网络结构聚氨酯-淀粉聚合物,其具有较高的透明度和拉伸强度,且当淀粉的分子量从924kDa降至269kDa时,聚合物的断裂伸长率从135%升至326%。Duarah等[22]以淀粉改性的多元醇合成出可生物降解超支化聚氨酯,其不仅具有较好的机械性能,还具有优异的形状记忆效应(人体温度下形状固定率可达98.8%,回复率达98.9%),且其机械性能在潮湿的环境中并没有较大的变化。Zhang等[23]用淀粉与聚己内酯聚氨酯预聚物在高速混炼机中进行充分混合反应制备淀粉-聚己内酯聚氨酯,此种方法可提高淀粉与聚己内酯的相容性,进而可提高聚氨酯的断裂伸长率。Biswas等[24]改变传统的热反应方法,采用微波工艺将淀粉与2, 4-二异氰酸甲苯酯反应制备聚氨酯,此种合成工艺节能,缩短了反应时间,且可取得和传统加热方法相同的转化率。
1.2.2 海藻酸基聚氨酯
海藻酸是一种亲水性的天然聚合物,可以从巨型海藻、海带、叶藻等中提取,盛产于各大洋海岸。海藻酸由均聚的α-L-吡喃古罗糖醛酸嵌段、均聚的β-D-吡喃甘露糖醛酸嵌段以及这两种糖醛酸的交替嵌段,以1, 4-糖苷键连接而成,且以钙、镁、钠、钾、锶盐等形式存在于许多海洋褐藻的细胞壁中。
迄今,海藻酸盐已广泛应用于食品、生物医药、纺织、印染等领域,其可制成微球、微胶囊、海绵、水凝胶、泡沫、弹性体、纤维等形式,增加了其在组织工程和药物释放等领域的应用。海藻酸中含有大量的羟基官能团,可以作为制备聚氨酯的材料,海藻酸基聚氨酯多以弹性体、纳米复合材料、水凝胶等产品形式广泛应用于食品和生物领域[25]。Wang等[26]通过自由基接枝聚合反应的方法制备了聚氨酯-海藻酸钙凝胶,聚氨酯侧链高度有序结晶的形成使海藻酸的结构发生重排,从而提高了此凝胶的热稳定性、结晶和抗膨胀性能。此凝胶的转印效率和可选择性随接枝率的增加而提高,研究人员利用此性能将其用于蛋白质识别,与单一的海藻酸钙相比,此凝胶具有较高的重新连接选择的能力,更易识别和分离目标蛋白分子[27]。尽管上述制备的聚氨酯-海藻酸凝胶在化学传感器和生物分离领域具有较好的应用前景,但其缺点是具有不同玻璃化转变温度的聚氨酯和海藻酸盐不相容,使凝胶易产生相分离。为提高聚氨酯和海藻酸盐的相容性,Daemi等[28]通过控制水性聚氨酯的离子性质制备了水性聚氨酯-海藻酸钠聚合物,动态热机械分析表明其只具有一个玻璃化转变温度和较优异的机械性能。他们[29]还通过将异氰酸酯预聚物与海藻酸盐反应制备了一种水溶性海藻酸基聚氨酯,并对其化学结构和反应率进行了研究。此外,采用溶液聚合的方法也可以解决聚氨酯与海藻酸盐的相容性问题,他们[30, 31]采用此方法将海藻酸钠和聚氨酯水分散液反应制备了具有较好相容性的聚氨酯-海藻酸纳米颗粒,此颗粒是一种具有较高弹性伸长率的热塑性弹性体。
1.2.3 甲壳素基聚氨酯
甲壳素是1823年由欧吉尔从甲壳动物外壳中提取出来的一种淡米黄色至白色的物质,其不溶于水、碱、一般的酸和有机溶剂、只溶于部分浓酸,甲壳素脱掉其中55%以上的N-乙酰基就成为可以溶于稀酸的壳聚糖。甲壳素和壳聚糖具有无毒、低免疫活性、可生物降解、抗菌和环境友好等优点[32~34]。因此,通过对甲壳素和壳聚糖进行化学改性得到的衍生物可望应用于生物医药、化妆品、食品、水处理等领域。目前已有多种基于甲壳素(壳聚糖)及其衍生物制备的功能材料,如抑菌膜、亲水膜、选择性蒸汽透过膜、微孔膜等。不溶不熔的甲壳素(壳聚糖)原材料难以处理和加工,且材料较脆,缺乏使用价值,而聚氨酯是一种具有优异性能的高分子材料,且是制备共混和互穿网络结构的理想材料,因此,可将甲壳素(壳聚糖)与聚氨酯采用共混或互穿网络方法制备甲壳素(壳聚糖)基聚氨酯复合材料。
Matsui等[35]将甲壳素与以聚己内酯为软段的聚氨酯采用直接共混的方法制备柔韧性好且易加工的甲壳素基聚合物。研究表明,在聚氨酯的玻璃化温度变化范围内,甲壳素和聚氨酯的相容性有很大程度的增加;当甲壳素结晶时,其相容性下降。随后,他们[36]研究了甲壳素和聚氨酯共混聚合物在生物领域的应用,其在生物体内无毒副作用且具有高稳定性。
Barikani等[37]采用壳聚糖和二羟甲基丙酸为扩链剂制备了壳聚糖基水性聚氨酯乳液,壳聚糖的加入提高了聚氨酯的热稳定性和表面疏水性能,但由于壳聚糖的加入量较少,其提高的程度有限。Zia等[38~40]以端羟基聚丁二烯为软段、甲壳素和1, 4-丁二醇为扩链剂制备了聚氨酯,并对其分子结构、表面形态和特性进行了研究,结果表明,甲壳素的加入使聚氨酯的吸水率和溶胀率降低,其抗菌性能随甲壳素含量的增加而增加。Saralegi等[41]将甲壳素以纳米晶体的形式在聚氨酯预聚体制备的过程中引入,该聚氨酯的软段和硬段都结晶使其具有形状记忆效应,而甲壳素的少量加入即可使聚氨酯硬段的结晶能力得到大幅增加,从而提高了聚氨酯的形状记忆效应。Chen等[42]以聚乙二醇为聚氨酯的亲水链段、甲壳素和异佛尔酮二异氰酸酯为亲油链段制备一种在极性有机溶剂中随温度变化可以转变为半固凝胶态的甲壳素基聚氨酯,并对其结构进行了表征。
2 导电聚氨酯
自2000年诺贝尔化学奖获得者Heeger、MacDiarmid和白川英树三位科学家通过研究证明了绝缘的高分子材料在一定的条件下也可以具有导电性后,导电高分子材料得到了迅速发展,已成为新兴基础有机功能材料之一。其主要包括共混型/掺杂型、结构型、高分子固体电解质和聚电解质四大类型,可应用于电极、抗静电、电容器、电磁屏蔽、传感器和生物材料等方面。聚氨酯自身不具有导电性,但其具有优异的物理和机械性能,以聚氨酯为基质制备导电材料,可以使材料不仅具有聚氨酯的良好性能,也同时具有导电性。导电聚氨酯的制备大多采用直接掺杂的方法,对于带有可反应官能团的导电材料也可采用原位聚合的方法制备。碳纳米管(CNT)和石墨烯以其特有的分子结构、小尺寸效应和优异的热学和电性能,已广泛应用于电极材料和纳米电子器件等多个领域[43~46]。
2.1 碳纳米管基导电聚氨酯
CNT是1991年日本的电子显微镜专家在检验石墨电弧设备中产生的球状碳分子时意外发现的,它是一种具有特殊结构和性能的纳米材料。CNT按石墨烯片的层数可分为:单壁碳纳米管(SWCNT)和多壁碳纳米管(MWCNT),与MWCNT相比,SWCNT直径大小的分布范围小,缺陷少,具有更高的均匀一致性。由于CNT中碳原子采取sp2杂化,s轨道成分比较大,使CNT具有较好的力学性能,其是目前可制备出的具有最高比强度的材料。CNT上碳原子的p电子可以形成大范围的离域π键,显著的共轭效应使其具有优异的导电性能。其次,CNT具有较高的导热率,复合材料中掺杂少量的CNT即可使其热导率得到很大改善。
Souri等[47]采用共混的方法制备了CNT基聚氨酯复合材料,并将其电性能与膨胀石墨基聚氨酯和杂化的MWCNT和石墨纳米片基聚氨酯进行比较。结果表明,当CNT加入量为5(wt)%时,CNT基聚氨酯具有最好的电性能,CNT加入量为7(wt)%时,其最高电导率达0.33S/m。将其作为电阻传感器对其性能进行测试,当CNT的加入量为5(wt)%时,其具有最好的电阻感应能力且可以承受2000个循环的重复测试。不过由于CNT在聚合物基质中很难分散,且其与基体的界面吸附强度很低,采取直接共混的方法使CNT的加入量有限,从而导致聚合物性能的提高受到一定的限制。Zeng等[48]将功能化的MWCNT与聚氨酯进行共价键结合,通过溶液共混的方法制备了一种质轻、柔韧且具有较好电磁屏蔽性能的MWCNT基水性聚氨酯复合薄膜(图 1)。当CNT的添加量为76(wt)%,聚氨酯薄膜的厚度为0.05、0.32和0.8mm时,其电磁屏蔽性能可分别达到24、49和80dB;其特异性电磁屏蔽性能最高可达3408dB cm2/g,远远高于其他的碳基复合材料。Jomaa等首先将CNT表面进行氧化引入羟基官能团,通过逐步反应将聚氨酯接枝到CNT表面,并对其电性能进行研究。结果表明,在CNT壁上接枝聚氨酯大分子降低了复合材料的导电性能,而将渗流阈值提高了5%,同时介电常数也得到大幅度提高。
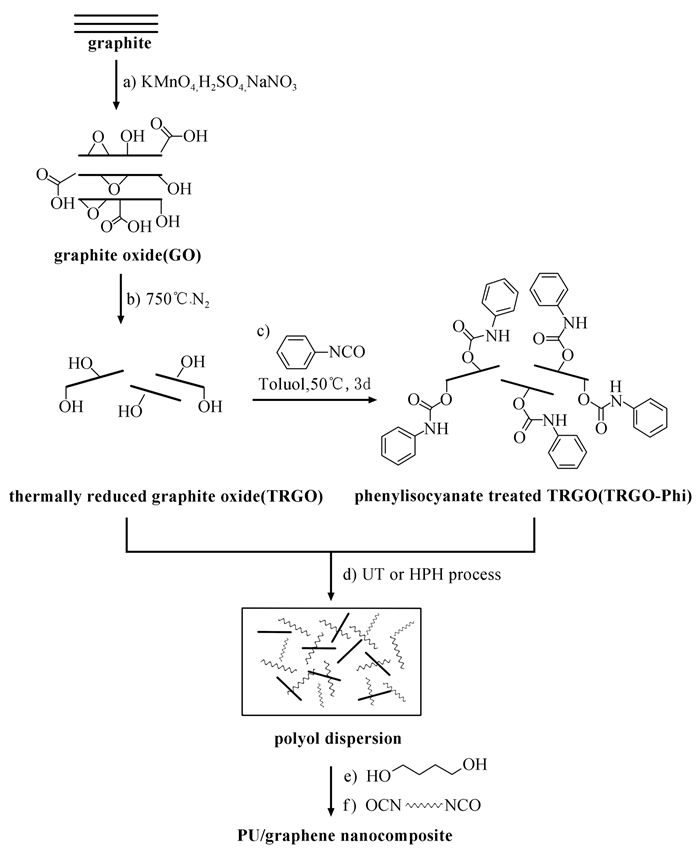
2.2 石墨烯基导电聚氨酯
石墨烯是由碳原子构成的新型二维纳米片层材料,2004年,英国物理学家Geim和Novoselov成功从石墨中分离出石墨烯,证明其可以单独存在。石墨烯分为单层、双层、少层、多层或厚层几种类型。石墨烯具有完美的二维晶体结构,它的晶格是由6个碳原子围成的六边形,厚度为一个原子层。碳原子之间由σ键连接,这些σ键赋予了石墨烯极其优异的力学性质和结构刚性。石墨烯的硬度比最好的钢铁强100倍,甚至还要超过钻石。在石墨烯中,每个碳原子都有一个未成键的p电子,这些p电子可以在晶体中自由移动,且运动速度高达光速的1/300,赋予了石墨烯良好的导电性。石墨烯是新一代的透明导电材料,在可见光区,四层石墨烯的透过率与传统的ITO薄膜相当,在其他波段,四层石墨烯的透过率远远高于ITO薄膜[49]。在塑料里掺入百分之一的石墨烯,就能使塑料具备良好的导电性;加入千分之一的石墨烯,能使塑料的抗热性能提高30摄氏度,在此基础上可以研制出薄、轻、拉伸性好和超强韧新型材料。在聚氨酯中引入石墨烯,可以实现聚氨酯在特殊功能方面的优异特性,也是石墨烯在实际应用中的一个重要发展方向。
石墨烯基聚氨酯的制备方法主要包括简单共混、原位聚合和接枝共聚方法。共混法制备工艺简单,但受石墨烯在聚氨酯基体中的分散性限制,使其整体性能的提高有限;通过将石墨烯与聚氨酯以化学键的方式制备复合材料是使两者优异性能得到较好发挥的有效途径(图式 4)[50]。Hodlur等[51]采用此方法合成了石墨烯在聚氨酯基体中可以均匀分布的压敏型复合导电材料,该复合材料在约50.7kPa的微弱压力下的电导率可提高超过5个数量级。
Huang等[52]采用自组装的方法制备了银纳米粒子-石墨烯-聚氨酯复合材料,此体系中,石墨烯作为连接银纳米粒子和聚氨酯的中间桥梁,当石墨烯和银纳米粒子(两者比例为5:1)添加的质量分数为0.05%时,复合材料的表面电阻达到150Ω/□,透光率达85%,且当将复合材料加热至100℃时,其透光率可进一步提高。当石墨烯和银纳米粒子的质量比为3:1时,复合材料具有较好的导电性和机械性能。Liao等[53]制备了具有低渗透阈值的石墨烯基聚氨酯复合导电材料,当石墨烯的添加量为0.15%时,其热稳定性明显增加,且材料自身形成导电网络结构,具有较好的导电性能。综上,将石墨烯与聚氨酯以化学键的形式结合,可以使石墨烯在体系中均匀分散形成较稳定的复合体系,是制备石墨烯基聚氨酯导电复合材料的较有效方法,具有较好的工业应用前景。
3 形状记忆聚氨酯
形状记忆高分子材料是一种可以对其施加一定的程序使其记忆一个临时形状,当对其进行适当的外界刺激,可以恢复到原始形状的智能材料。常见的具有形状记忆的高分子材料是对温度刺激响应的材料,将其加热至热转变温度(如玻璃化转变温度或结晶熔融温度)以上,同时施加一定的外力使其变形为一个临时形状,降温至热转变温度以下,此临时形状被固定;然后撤去外力,在将其重新加热至热转变温度以上时,其可以恢复到原始形状[54]。目前,形状记忆高分子材料主要包括单向形状记忆和双向(可逆)形状记忆两大类。单向形状记忆体系在加热时可以恢复到原始形状,然而在冷却时却不能恢复其低温下的临时形状。具有单向形状记忆效应的高分子材料形变简单,已在传感器、制动器、航空航天、生物医学等领域得到实际应用[55, 56],但其单向形状记忆行为却在一定程度上阻碍了形状记忆高分子的应用范围和发展前景。双向形状记忆体系在加热时可以恢复到原始形状,在冷却时也可恢复其低温下的临时形状,此可逆形状记忆行为大大提高了形状记忆高分子材料的应用领域,是目前形状记忆材料的重要研究方向[57~59]。
聚氨酯独特的性能使其成为制备形状记忆高分子的理想材料。Zotzmann等[60, 61]采用结晶性聚己内酯和聚十五酸内酯与脂肪族二异氰酸酯反应制备出软段含有两个结晶链段的聚氨酯交联体系。此材料在室温或在其结晶熔融温度之上的断裂伸长率可达250%,聚氨酯中聚己内酯的含量为50(wt)%时,其在结晶温度之下的形状变形固定率达80%,加热至结晶温度之上,其原始形状回复率达97%。随后他们还研究了此体系在外界力作用下的双向形状记忆行为。
2010年,胡金莲课题组制备出一种具有双向形状记忆效应的双层结构聚氨酯体系。他们将具有形状记忆效应的聚氨酯薄膜(命名为层0)加热至其热转变温度之上,对其进行预拉伸,并将温度降至室温,使预拉伸后的形状固定(命名为层1),再使用胶粘剂将层1与另一种聚氨酯弹性薄膜(命名为层2)进行粘结,制备出一种双层结构的复合聚氨酯体系。此复合体系中具有形状记忆效应的层1在加热至其热转变温度以上会进行形状恢复,但由于受层2的限制,层1的形状恢复使复合体系产生向层1的弯曲变形。当将温度降低至形状记忆层的热转变温度之下时,复合体系的弯曲度降低。此种体系在不需要外力作用的条件下可以在弯曲和逆弯曲两种形状之间进行可逆变化[62]。Xie[63]对此体系的工作机理进行了深入研究,他指出此体系与早期液晶高分子通过施加恒重产生的冷伸长和热收缩可逆形变是相似的,此体系是将形状记忆层在其形状恢复后与弹性层在形状上的不匹配产生的内应力代替了施加恒重产生的外力作用。此体系的提出虽然是双向形状记忆高分子的一个重要进步,但其具有的缺点是体系产生的内应力使其只能在两种固定的形状之间进行可逆变化。
2013年,Behl等[64]研究了Zotzmann等制备的含有两个结晶链段的聚氨酯交联体系的双向形状记忆行为,通过对体系进行程序化定型,实现了高分子材料在无外力作用下的双向形状记忆行为(见图 2)。此项研究是双向形状记忆高分子的一个重要突破,目前只有这个体系可以实现程序化的定形。但该体系高温结晶相的热转变温度不易调控,一方面由于高温结晶相原材料的选择种类有限,另一方面只能加入少量共聚单体以实现小范围的调控,如果共聚单体加入太多会破坏其结晶,因此大大限制了双向形状记忆高分子的应用范围。另外,结晶相的结晶度对热机械条件比较敏感,不适宜用于对双向形状记忆高分子机理的定量研究。
4 机械响应聚氨酯
传统意义上的机械响应变色聚合物材料是一类在外力(如拉力、压力、剪切力等)作用下引发化学反应致使其颜色发生明显改变的力刺激响应型智能材料,其在微应力传感、信息存储、商标防伪和发光器件等领域具有重要的潜在应用[65~68]。实现材料力致变色的途径有两种:一是外力作用下使材料的分子化学结构发生改变;二是改变材料的聚集态结构。其中通过外力作用改变分子的化学结构,利用材料受力前后不同的分子结构发出不同颜色的光来实现机械响应变色是最容易实现的方法[69]。然而,在固体状态下,通过外界力刺激实现材料分子水平化学结构的改变非常困难,目前,机械响应变色聚合物材料最典型的结构是含螺吡喃结构单元的高分子,其可在外力作用下由无色态的闭环体螺吡喃转变为有色态的开环体部花菁结构[69~72]。
螺吡喃的分子结构类似于螺噁嗪,是一类研究最早、最广泛的有机光致变色分子,其热致变色性质早在1921年就已被发现,但直到1952年Fisher和Hirsherg才发现它的光致变色性质。螺吡喃的机械响应特性直到近年来才被广泛研究和应用,其作为一种无损探伤手段,可广泛应用于对各种物体的涂层、桥梁、机翼甚至起重行业的钢丝绳进行检测,以维护高危险度作业的安全。Davis等[73]合成了一系列螺吡喃衍生物,并研究了其在外力作用下的变色行为,证明了通过对螺吡喃分子进行合理的结构设计,并将其引入到聚丙烯酸酯主链体系可以制备出具有机械响应的智能高分子材料。此类体系的原理是通过共价键将螺吡喃引入到聚合物主链中,在一定的外力作用下,聚合物中心共价的螺碳氧键断裂,其分子扭曲为平面共轭形式,由闭环体螺吡喃转变为开环体部花菁结构。随后O′Bryan等[74]合成出螺吡喃分子中螺环两侧分别含有羟基的单体,将其与己内酯在辛酸亚锡催化剂的作用下开环聚合制备出颜色随外力发生明显变化的聚己内酯,同时他们利用密度泛函理论(DFT)和时间依赖的密度泛函理论(TD-DFT)证实了此类聚合物的颜色变化是由螺环上的碳氧键在外力作用下断裂生成了有色的部花菁结构引起的。
机械响应聚合物材料在力作用下的颜色变化受多种因素的影响。内部因素有螺吡喃的引入方式、螺吡喃单元的引入数量、螺吡喃的结构、螺吡喃在分子链上的位置、聚合物链的定向与松弛、聚合物的分子量和聚合度[75, 76];外部因素有温度、力的施加模式、力的施加速率、力的施加时间等[77~79]。将螺吡喃分子通过螺环两侧的官能团以共价键形式引入到聚丙烯酸酯、聚己内酯等聚合物链中,可以制备出一类新颖的机械响应变色聚合物材料,但由于聚丙烯酸甲酯柔韧性高、玻璃化温度低,而聚甲基丙烯酸甲酯玻璃化温度高、柔韧性差,聚己内酯因其结构的柔软性导致机械强度差,其都不适合用于室温下研究固体聚合物在外力作用下分子链中螺吡喃的动力学和热力学转变行为,不适合作为机械响应聚合物进行定量研究[72]。
聚氨酯材料自身具有柔韧性、弹性和较低的玻璃化转变温度,是研究室温下螺吡喃在外力作用下结构变化的理想材料。Lee等[81]采用逐步聚合工艺制备了螺吡喃-聚氨酯体系(图 3),通过光谱分析研究了室温条件下体系在外力作用下螺吡喃中螺环上较弱碳氧键断裂生成有色态部花菁的动力学过程,并研究了其由部花菁结构转变为螺吡喃结构的热力学性质。Zhang等[82]以聚四氢呋喃为软段、2-脲基-4[1H]-嘧啶酮(UPy)为硬段将螺吡喃同时以化学和物理交联的方法引入到聚氨酯体系中,研究了聚氨酯材料在外力作用下的断裂情况和颜色变化的关系,并提出螺吡喃有望作为“分子探针”感应高分子材料被拉伸时内部的受力情况。
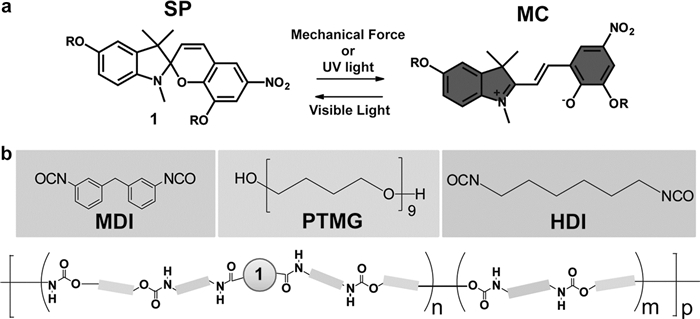 图3
(a) 螺吡喃单体受力或紫外/可见光照射后结构变化示意图;(b)螺吡喃-聚氨酯合成路线图[81]
Figure3.
(a) Chemical structures of spiropyran (SP) and merocyanine (MC) and the mechanically or optically triggered conversion equilibrium between the colorless SP and colored MC forms. (b) Schematic of the incorporation of SP mechanophore 1 into PU via step growth polymerization[81]
图3
(a) 螺吡喃单体受力或紫外/可见光照射后结构变化示意图;(b)螺吡喃-聚氨酯合成路线图[81]
Figure3.
(a) Chemical structures of spiropyran (SP) and merocyanine (MC) and the mechanically or optically triggered conversion equilibrium between the colorless SP and colored MC forms. (b) Schematic of the incorporation of SP mechanophore 1 into PU via step growth polymerization[81]
从响应机理定量研究的角度上,由于机械响应聚合物材料在力作用下的颜色变化受材料内部结构、外部因素等多种因素的影响,而聚氨酯原材料结构多样,可以根据性能对其结构进行合理的设计,同时聚氨酯在不同温度下的机械性能可进行程序化的调控,此外,其优越的的加工性、可多功能化及大形变都是其他聚合物材料所不可比拟的。根据机械响应聚合物材料受力前后的颜色变化,能够反映出材料的损伤程度,其可以作为一种无损探伤手段应用到很多场合。基于聚氨酯的机械响应材料可以为无损探伤这方面的未来研究及应用开发提供一个全新的方向。
5 结语
传统的聚氨酯以其优异的性能广泛应用于制造塑料、橡胶、纤维、硬质和软质泡沫塑料、胶粘剂和涂料等多种材料。近年来,高分子材料逐渐向智能化、多样化、环境友好等方向发展,聚氨酯材料因其分子结构设计的可控、原材料种类多样等优点,使其在功能高分子材料领域占有重要的地位。石油基聚氨酯的原材料多为不可再生资源,且多有毒有害,因此用生物质产品取代石油原材料制备绿色环保型聚氨酯材料是今后社会发展的需要。但目前生物基聚氨酯产品的性能还无法与石油基聚氨酯相媲美,如何通过新的合成手段去实现生物基聚氨酯结构与性能的可控是未来研究和发展的方向。以聚氨酯为基质制备的可导电聚氨酯材料同时具有聚氨酯的优异性能和良好的导电性,是导电材料迈向实际应用的重要研究方向。目前制备导电型聚氨酯的主要方法为共混、接枝共聚和原位聚合。其中,共混法操作简单,但导电材料在体系的分散性问题和与聚氨酯基体间的相互作用力弱等问题需要解决;接枝共聚和原位聚合可以使导电材料和聚氨酯形成强的化学键合作用,具有较好的工业前景。形状记忆聚氨酯材料是一种具有形状记忆效应的新型智能高分子材料,但目前很多形状记忆聚氨酯材料存在形状回复力弱、回复速度慢、回复精确度低、可逆形状记忆效果差且物理机械性能较差等缺点,因此,如何通过合理的结构设计及化学改性制备具有较好形状记忆行为的聚氨酯材料是今后研究的热点。聚氨酯材料自身的柔韧性、弹性和较低的玻璃化转变温度是制备和研究室温下螺吡喃在外力作用下发生结构变化的理想材料。目前对机械响应型聚氨酯材料的研究多在初步阶段,对其响应机理进行定量研究并在此基础上开发出基于机械响应聚氨酯的智能材料作为一种无损探伤手段,具有较好的应用前景。
-
-
[1]
P H Chen, Y F Yang, D K Lee et al. Adv. Polym. Technol., 2007, 26(1):33~40. doi: 10.1002/(ISSN)1098-2329
-
[2]
M A Hood, B B Wang. Polymer, 2010, 51(10):2191~2198. doi: 10.1016/j.polymer.2010.03.027
-
[3]
D K Chattopadhyay, K V S N Raju. Prog. Polym. Sci., 2007, 32(3):352~418. doi: 10.1016/j.progpolymsci.2006.05.003
-
[4]
C H Tsou, H T Lee, H A Tsai et al. Polym. Degrad. Stabil., 2013, 98(2):643~650. doi: 10.1016/j.polymdegradstab.2012.11.010
-
[5]
L Poussard, F Burel, J P Couvercelle et al. Biomaterials, 2004, 25(17):3473~3483. doi: 10.1016/j.biomaterials.2003.10.069
-
[6]
A Noreen, K M Zia, M Zuber et al. Prog. Org. Coat., 2016, 91:25~32. doi: 10.1016/j.porgcoat.2015.11.018
-
[7]
H Mutlu, M A R Meier. Eur. J. Lipid Sci. Tech., 2010, 112(1):10~30. doi: 10.1002/ejlt.v112:1
-
[8]
张欢, 周建军, 何明等.材料导报A, 2014, 28(12):91~95. http://www.cqvip.com/QK/90370X/201423/663390169.html
-
[9]
S Miao, P Wang, Z Su et al. Acta Biomater., 2014, 14(4):1692~1704.
-
[10]
L Hojabri, X Kong, S S Narine. Biomacromolecules, 2009, 10(4):884~891. doi: 10.1021/bm801411w
-
[11]
L Hojabri, X Kong S S Narine. J. Polym. Sci. Polym. Chem., 2010, 48(15):3302~3310. doi: 10.1002/pola.24114
-
[12]
L Hojabri, X Kong S S Narine. Biomacromolecules, 2010, 11(4):911~918. doi: 10.1021/bm901308c
-
[13]
S Miao, L Su, P Wang et al. Eur. J. Lipid Sci. Technol., 2012, 114(10):1165~1174. doi: 10.1002/ejlt.v114.10
-
[14]
A B Chaudhari, A Anand, S D Rajput et al. Prog. Org. Coat., 2013, 76(12):1779~1785. doi: 10.1016/j.porgcoat.2013.05.016
-
[15]
C E Hoyle, C N Bowman. Angew. Chem. Int. Ed., 2010, 49(9):1540~1573. doi: 10.1002/anie.200903924
-
[16]
C Q Fu, J C Liu, H Y Xia et al. Prog. Org. Coat., 2015, 83:19~25. doi: 10.1016/j.porgcoat.2015.01.020
-
[17]
G M Wu, Z W Kong, H Huang et al. J. Appl. Polym. Sci., 2009, 113(5):2894~2901. doi: 10.1002/app.v113:5
-
[18]
F Zia, K M Zia, S Kamal et al. Carbohyd. Polym., 2015, 134:784~798. doi: 10.1016/j.carbpol.2015.08.034
-
[19]
D Y Yang, H Q Zhang, X S Rong et al. Plast. Rubber Compos., 2012, 41(10):425~429. doi: 10.1179/1743289812Y.0000000009
-
[20]
S J Lee, B K Kim. Carbohyd. Polym., 2012, 87(2):1803~1809. doi: 10.1016/j.carbpol.2011.09.098
-
[21]
X D Cao, Y Z Tao, L A Lucia et al. J. Appl. Polym. Sci., 2010, 116(3):1299~1305. doi: 10.1002/app.31497
-
[22]
R Duarah, Y P Singh, B B Mandal et al. New J. Chem., 2016, 40(6):5152~5163. doi: 10.1039/C5NJ03294F
-
[23]
X Zhang, Y Zhang, J J Liao et al. J. Appl. Polym. Sci., 2015, 132(32):1~10. doi: 10.1002/app.42381/pdf
-
[24]
A Biswas, S Kim, Z Q He et al. Int. J. Polym. Anal. Charact., 2015, 20(1):1~9. doi: 10.1080/1023666X.2015.975017
-
[25]
K M Zia, F Zia, M Zuber et al. Int. J. Biol. Macromol., 2015, 79:377~387. doi: 10.1016/j.ijbiomac.2015.04.076
-
[26]
J C Wang, X G Ying, X Li et al. Mater. Lett., 2014, 126:263~266. doi: 10.1016/j.matlet.2014.03.178
-
[27]
L X Li, X G Ying, J Q Liu et al. Mater. Lett., 2015, 143:248~251. doi: 10.1016/j.matlet.2014.12.108
-
[28]
H Daemi, M Barikani, M Barmar. Int. J. Biol. Macromol., 2014, 66(3):212~220.
-
[29]
H Daemi, M Barikani. Carbohyd. Polym., 2014, 112(21):638~647.
-
[30]
H Daemi, M Barikani, M Barmar. Carbohyd. Polym., 2013, 92(1):490~496. doi: 10.1016/j.carbpol.2012.09.046
-
[31]
H Daemi, M Barikani, M Barmar. Carbohyd. Polym., 2013, 95(2):630~636. doi: 10.1016/j.carbpol.2013.03.039
-
[32]
J Kumirska, M X Weinhold, J Thöming et al. Polymers, 2011, 31(4):1875~1901. http://citeseerx.ist.psu.edu/viewdoc/summary?doi=10.1.1.393.9587
-
[33]
V K Mourya, N N Inamdar. React. Funct. Polym., 2008, 68(6):1013~1051. doi: 10.1016/j.reactfunctpolym.2008.03.002
-
[34]
A Usman, K M Zia, M Zuber et al. Int. J. Biol. Macromol., 2016, 86:630~645. doi: 10.1016/j.ijbiomac.2016.02.004
-
[35]
M Matsui, M Munaro, L Akcelrud. Polym. Int., 2010, 59(8):1090~1098. doi: 10.1002/pi.2833/full
-
[36]
M Matsui, L Ono, L Akcelrud. Polym. Test., 2012, 31(1):191~196. doi: 10.1016/j.polymertesting.2011.09.006
-
[37]
M Barikani, H Honarkar, M Barikani. Monatsh. Chem., 2010, 141(6):653~659. doi: 10.1007/s00706-010-0309-1
-
[38]
K M Zia, K Mahmood, M Zuber et al. Int. J. Biol. Macromol., 2013, 59(4):320~327. http://www.ncbi.nlm.nih.gov/pubmed/23643975
-
[39]
K M Zia, N A Qureshi, M Mujahid et al. Int. J. Biol. Macromol., 2013, 59(4):313~319. http://www.ncbi.nlm.nih.gov/pubmed/23603077
-
[40]
K M Zia, M Zuber, M J Saif et al. Int. J. Biol. Macromol., 2013, 62(62):670~676. http://www.ncbi.nlm.nih.gov/pubmed/24120963
-
[41]
A Saralegi, S C M Fernandes, A A Varona et al. Biomacromolecules, 2013, 14(12):4475~4482. doi: 10.1021/bm401385c
-
[42]
S H Chen, C T Tsao, C H Chang et al. Carbohyd. Polym., 2012, 88(4):1483~1487. doi: 10.1016/j.carbpol.2012.01.055
-
[43]
Z Wu, Z Chen, X Du et al. Science, 2004, 305, 1273~1276. doi: 10.1126/science.1101243
-
[44]
R H Baughman, A A Zakhidov, W A D Heer. Science, 2002, 297(5582):787~792. doi: 10.1126/science.1060928
-
[45]
E Pop, D Mann, Q Wang et al. Nano. Lett., 2006, 6(1):96~100. doi: 10.1021/nl052145f
-
[46]
G Begtrup, K Ray, B Kessler et al. Phys. Rev. Lett., 2007, 99(23):155901~155904.
-
[47]
H Souri, I W Nam, H K Lee. Compos. Sci. Technol., 2015, 121(14):41~48.
-
[48]
Z H Zeng, M J Chen, H Jin et al. Carbon, 2016, 96:768~777. doi: 10.1016/j.carbon.2015.10.004
-
[49]
C Lee, X Wei, J W Kysar et al. Science, 2008, 321(5887):385~388. doi: 10.1126/science.1157996
-
[50]
A K Appel, R Thomann, R Mülhaupt. Polymer, 2012, 53(22):4931~4939. doi: 10.1016/j.polymer.2012.09.016
-
[51]
R M Hodlur, M K Rabinal. Compos. Sci. Technol., 2014, 90(90):160~165. doi: 10.1016/j.compscitech.2013.11.005
-
[52]
Y L Huang, A Baji, H W Tien et al. Carbon, 2012, 50(10):3473~3481. doi: 10.1016/j.carbon.2012.03.013
-
[53]
K H Liao, Y T Park, A Abdala et al. Polymer, 2013, 54(17):4555~4559. doi: 10.1016/j.polymer.2013.06.032
-
[54]
J S Leng, X Lan, Y J Liu et al. Prog. Mater. Sci., 2011, 56(7):1077~1135. doi: 10.1016/j.pmatsci.2011.03.001
-
[55]
李兴建, 王亚茹, 郑朝晖等.化学进展, 2013, 25(10):1726~1738. http://www.cqvip.com/QK/98085X/201310/47286079.html
-
[56]
朱光明, 魏堃, 王坤.高分子材料科学与工程, 2013, 26(8):168~171. http://www.cqvip.com/qk/94461x/201008/35020899.html
-
[57]
S Chen, J Hu, H Zhao. Compos. Sci. Technol., 2010, 70(10):1437~1443. doi: 10.1016/j.compscitech.2010.01.017
-
[58]
S A Turner, J Zhou, S S Sheiko et al. ACS Appl. Mater. Interf., 2014, 6(11):8017~8021. doi: 10.1021/am501970d
-
[59]
J J Li, R William, T Xie. Polymer, 2011, 52(23):5320~5325. doi: 10.1016/j.polymer.2011.09.030
-
[60]
J Zotzmann, M Behl, Y K Feng et al. Adv. Funct. Mater., 2010, 20(20):3583~3594. doi: 10.1002/adfm.v20:20
-
[61]
J Zotzmann, M Behl, D Hofmann et al. Adv. Mater., 2010, 22(31):3424~3429. doi: 10.1002/adma.200904202
-
[62]
S Chen, J Hu, H Zhao. Composi. Sci. Technol., 2010, 70(10):1437~1443. doi: 10.1016/j.compscitech.2010.01.017
-
[63]
T Xie. Polymer, 2011, 52(22):4985~5000. doi: 10.1016/j.polymer.2011.08.003
-
[64]
M Behl, K Kratz, J Zotzmann et al. Adv. Mater., 2013, 25(32):4466~4469. doi: 10.1002/adma.v25.32
-
[65]
S S Zeng, D Y Zhang, W H Huang et al. Nat. Chem., 2016, 7:11802. doi: 10.1186/1479-5876-11-142
-
[66]
Z Chi, X Zhang, B Xu et al. Chem. Soc. Rev., 2012, 41(31):3878~3896.
-
[67]
Y Sagara, T Kato. Nat. Chem., 2009, 1(8):605~610. doi: 10.1038/nchem.411
-
[68]
F Ciardelli, G Ruggeri, A Pucc. Chem. Soc. Rev., 2013, 42(3):857~870. doi: 10.1039/C2CS35414D
-
[69]
S L Potisek, D A Davis, N R Sottos et al. J. Am. Chem. Soc., 2007, 129(45):13808~13809. doi: 10.1021/ja076189x
-
[70]
刘水平, 张瑜, 刘峻等.东华大学学报(自然科学版), 2008, 34(6):647~651. http://www.cqvip.com/Main/Detail.aspx?id=29149741
-
[71]
姚献东, 池振国.中国科学:化学, 2013, 43(9):1090~1104.
-
[72]
张国峰, 陈涛, 李冲等.有机化学, 2013, 33(5):927~942. http://www.cqvip.com/QK/93463X/201305/45903556.html
-
[73]
D A Davis, A Hamilton, J Yang et al. Nature, 2009, 459(7243):68~72. doi: 10.1038/nature07970
-
[74]
G O'Bryan, B M Wong, J R McElhanon. ACS Appl. Mater. Interf., 2010, 6(2):1594~1600. http://www.ncbi.nlm.nih.gov/pubmed/20568704
-
[75]
B A Beiermann, S L B Kramer, J S Moore et al. ACS Macro Lett., 2012, 1(1):163~166. doi: 10.1021/mz2000847
-
[76]
B A Beiermann, S L B Kramer, P A May et al. Adv. Funct. Mater., 2014, 24(11):1529~1537. doi: 10.1002/adfm.v24.11
-
[77]
P A May, N F Munaretto, M B Hamoy et al. ACS Macro Lett., 2016, 5(2):177~180. doi: 10.1021/acsmacrolett.5b00855
-
[78]
G R Gossweiler, T B Kouznetsova, S L Craig. J. Am. Chem. Soc., 2015, 137(19):6148~6151. doi: 10.1021/jacs.5b02492
-
[79]
J W Kim, Y Jung, G W Coates et al. Macromolecules, 2015, 48(5):1335~1342. doi: 10.1021/ma502555d
-
[80]
C M Degen, P A May, J S Moore et al. Macromolecules, 2013, 46(46):8917~8921.
-
[81]
C K Lee, D A Davis, S R White et al. J. Am. Chem. Soc., 2010, 132(45):16107~16111. doi: 10.1021/ja106332g
-
[82]
H Zhang, Y J Chen, Y J Lin. Macromolecules, 2014, 47(19):6783~6790. doi: 10.1021/ma500760p
-
[1]
-



图 3 (a) 螺吡喃单体受力或紫外/可见光照射后结构变化示意图;(b)螺吡喃-聚氨酯合成路线图[81]
Figure 3 (a) Chemical structures of spiropyran (SP) and merocyanine (MC) and the mechanically or optically triggered conversion equilibrium between the colorless SP and colored MC forms. (b) Schematic of the incorporation of SP mechanophore 1 into PU via step growth polymerization[81]
-



 下载:
下载:





 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 0
- HTML全文浏览量: 0
 下载:
下载:
