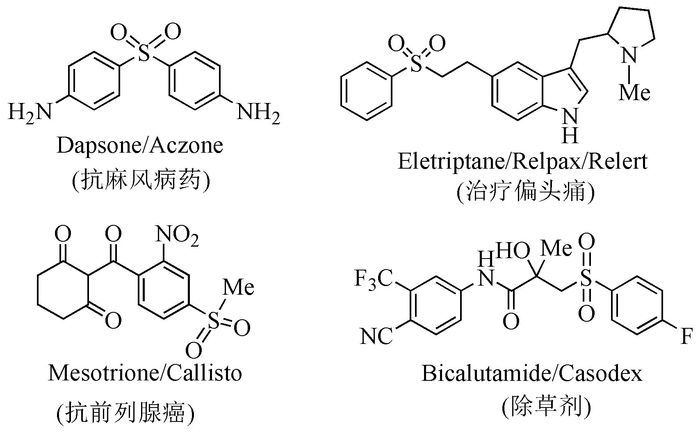
 图式 1
含有芳基砜的代表性药物和农药
Scheme1.
Typical drugs and pesticide containing aryl sulfone motif
图式 1
含有芳基砜的代表性药物和农药
Scheme1.
Typical drugs and pesticide containing aryl sulfone motif
引用本文:
肖彩琴. 砜类化合物合成研究进展[J]. 化学通报,
2017, 80(10): 908-917.

Citation: Xiao Caiqin. Progress in the Syntheses of Sulfones[J]. Chemistry, 2017, 80(10): 908-917.
Citation: Xiao Caiqin. Progress in the Syntheses of Sulfones[J]. Chemistry, 2017, 80(10): 908-917.
砜类化合物合成研究进展
English
Progress in the Syntheses of Sulfones
Abstract:
The broad spectrum of biological activity of sulfone compounds endows a wide applications in chemistry, medicine, agrochemistry and material science.Being an strong electron-withdrawing group, elongation of carbon chain could easily achieved via reaction of aliphatic sulfones with various electrophiles under basic condition.As a result, novel synthetic methodologies have continuous been developed especially during the past decades.This paper aimed at summarizing the synthetic methods of the sulfone compounds appeared in recent years, emphasizing the oxidation of sulfides, sulfonylation with sulfonyl chlorides and coupling of sulfinic acid salts.
-
砜是一类重要的有机合成中间体, 在化学、医药、农药及材料科学[1~4]合成中有着广泛的应用, 且具有一定的生物活性。例如, 多种芳基砜表现出良好的抗真菌[5]、抗菌[6]、抗癌[7]、抗艾滋病[8]、抗麻风病[9]、抗利什曼[10]等药理活性(图式 1);在COX-2抑制剂中也发现了芳基砜[11];此外, 最近的研究发现, α, β-不饱和砜可作为诱导VCAM-1(血管细胞粘附分子-1)的抑制剂[12];乙烯基砜则是多种新型染料的中间体[13]。还有, 砜官能团易于在温和的条件下引入和脱除, 砜基的引入可以活化α-位碳原子, 使其作为反应中心与酸和各种亲电试剂(如卤代烃、醛等)发生反应, 还可以用于C-C键的形成[14], 在有机合成领域具有极高的应用价值。
图式 1
含有芳基砜的代表性药物和农药
Scheme1.
Typical drugs and pesticide containing aryl sulfone motif
已报道的砜的合成方法主要有硫醚氧化、二氧化硫加成、傅克磺酰化和过渡金属催化偶联等。本文主要介绍近几年有机砜类化合物的合成方法研究进展。
1 硫醚的氧化
在砜类化合物的众多合成方法中,硫醚氧化是最直接的一种方法,常用的氧化剂有金属氧化物、无机和有机氧化剂等。另外使用诸如O2、HNO3、H2O2、N2O4、MnO2、高碘酸等其他氧化剂的结果也有报道。但如何将硫醚选择性地氧化成亚砜或砜是多年来一直为广大学者研究的重点。
1995年,Aldea等[15]在用钯的配合物[Pd(PtBu2H)(μ-PtBu2)]2作催化剂、O2作氧化剂,将脂肪族、芳香族、烯丙基、苄基和杂环类硫醚选择性氧化成亚砜的过程中发现,向该催化体系中加入BPPM[(2S, 4S)-4-二苯基膦-2-(二苯基膦甲基)-N-叔丁氧羰基吡咯烷]配体时,产物中砜和亚砜以2.4:1存在于反应体系中(式(1)),由此可知,对于氧化性较弱的催化体系,适当配体的存在对硫醚的氧化起着关键作用。
随后,Smart等[16]发现HOF-CH3CN复合物是一种简单易得的良好氧化剂,可被用在脂肪烃、芳香烃、羟基、羰基和胺基的氧化反应中。受此启发,他们尝试将该复合物用在硫醚氧化反应中,在过量HOF-CH3CN复合物存在的条件下,室温反应20min,几乎完全定量地得到相应的砜类化合物;尤其是,一些连有吸电子基团的烷烃、芳香烃类的硫醚亦可被氧化成砜。可见,HOF-CH3CN复合物表现出了非凡的氧化活性。
1997年,Varma等[17]在无溶剂条件下,通过控制硫化物与SiO2负载的高碘酸物料比选择性地得到相应的亚砜或砜。当二者比例为1:1.7时,微波辐射30~150 s后,可以较高产率得到亚砜;比例为1:3时,可快速、高产率得到砜(72%~93%)。该方法不仅适用于芳烃类砜的合成,同时也适用于溶解度较差、通常不易氧化的长链脂肪烃类砜的合成。对于二苯基噻吩上的硫原子,也可在以上反应条件下直接被氧化成砜,由此可见,该氧化体系可将杂环上的硫原子成功氧化(式(2))。
尽管以上氧化体系均可较容易地将硫醚氧化成砜,但遗憾的是他们均未对带有易氧化性官能团的底物进行探索,故筛选出一种对易氧化性官能团具有良好兼容性的氧化体系也是一种新的挑战。2003年,Xu等[18]发现,以H5IO6/CrO3为氧化体系,硫醚上无论是连有吸电子基团还是易氧化的双键、叁键、-OH、-CHO、-NH2等官能团时,以乙腈为溶剂,室温下,均可高产率(90%~97%)、高选择性地将其氧化为相应的砜。该氧化体系对易氧化的官能团表现出了良好的兼容性,例如,含硫杂环类的硫醚可以被高选择性地氧化而不影响杂环硫原子(图式 2)。但该反应中有毒试剂的使用可能会限制其在实际生产中的应用。
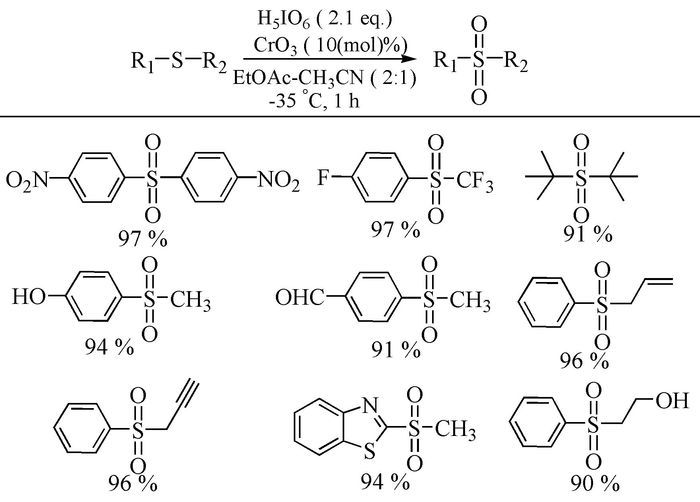 图式 2
H5IO6/CrO3选择性将硫醚氧化成砜
Scheme2.
H5IO6/CrO3 chemoselective oxidation of sulfides to sulfones
图式 2
H5IO6/CrO3选择性将硫醚氧化成砜
Scheme2.
H5IO6/CrO3 chemoselective oxidation of sulfides to sulfones
在普遍提倡环境保护、绿色生产、讲究原子经济性的今天,寻找一种绿色、环保、高效的氧化体系仍然是该类反应今后发展的主要趋势。绿色氧化剂H2O2价廉、反应后处理简单、副产物为水,因而受到化学工作者广泛关注。围绕该类氧化体系,人们展开了大量的研究,以H2O2为氧化剂的多种催化体系如离子液体、钨催化体系、钼催化体系、钒催化体系等陆续被报道[19]。2006年Bahrami等[20]发现,在甲醇溶液中,将硫化物/H2O2/ZrCl4以摩尔比2:20:5混合,室温下反应1~4 min,几乎可以使硫化物定量转化为砜(95%~99%),同时,对卤素、硝基及双键表现出较好的兼容性,从而实现了该类反应的绿色合成(式(3))。
Rahimizadeh等[21]以纳米材料修饰过的TiO2为催化剂、30% H2O2为氧化剂,硫醚可在室温条件下被高选择性地氧化为砜,产率可达100%。该方法同时适用于带有双键、氰基和羟基等易氧化官能团的芳烃和脂肪烃类硫化物的氧化,具有选择性高、氧化条件温和且催化剂能重复利用等优点(式(4))。
2010年,Rostam等[22]在无溶剂条件下,向相对不够活泼的二苯基硫及其衍生物中加入过量(3.6倍量)的30% H2O2和少量硼酸,室温下搅拌25min,可高产率得到砜类化合物。而且,该反应对官能团具有较好的耐受性,例如,二苯基硫上连有的-OH、-CHO等易氧化的官能团不受影响(图式 3)。
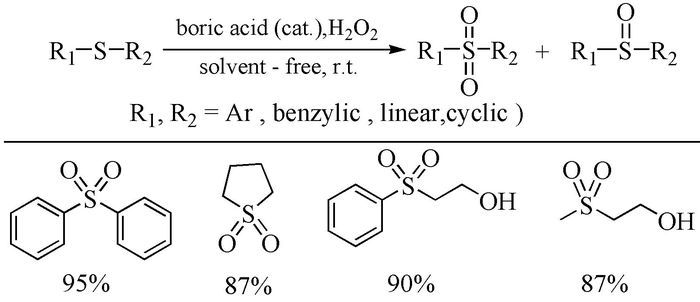 图式 3
无溶剂条件下用H2O2和硼酸作催化剂的硫醚氧化反应
Scheme3.
A oxidation of sulfides using hydrogen peroxide and boric acid as the catalyst under solvent-free conditions
图式 3
无溶剂条件下用H2O2和硼酸作催化剂的硫醚氧化反应
Scheme3.
A oxidation of sulfides using hydrogen peroxide and boric acid as the catalyst under solvent-free conditions
2012年,Jereb等[23]发现,在无溶剂和催化剂的条件下,75℃时,用30% H2O2作氧化剂可将硫化物以较高产率(41%~95%)氧化成砜,与前面的几种方法相比较,该方法具有原子经济性高、副产物少、污染小等优点(式(5))。
2014年,Maleki等[24]报道了用二氧化硅的溴化物为催化剂、30% H2O2为氧化剂、乙腈作溶剂,可将硫化物选择性地氧化为亚砜或砜。同时,Chakravarthy等[25]发现了一种基于表面活性剂的钼系催化剂,在水的介质中,它表现出了较强的氧化性,可将硫化物氧化为砜。
随着人们研究的深入,虽然H2O2氧化硫醚合成砜的反应得到了发展,尤其是一系列新催化剂的应用使该类化合物的合成反应条件日臻完善。但H2O2因其中等的氧化能力,在完成高选择性、高效的硫醚氧化反应时,大多需加入金属催化剂和有机溶剂,对环境或多或少存在一定的污染。
2 磺酰氯参与的磺酰化反应
传统Friedel-Crafts磺酰化反应多以磺酰氯、磺酸或磺酸盐为酰化试剂,在Lewis酸或质子酸的催化下,与卤代烃、芳烃或有机硼酸等发生亲电取代来制备砜类化合物。多年来,寻找一种廉价易得、绿色高效的催化剂成为人们不懈追求的目标之一。Friedel-Crafts磺酰化中最典型的催化剂为AlCl3和BF3[26],还有后来被报道的InF3[27]、三氟甲磺酸/BiCl3及有趣的新催化剂Zn-交换的分子筛、Fe(Ⅲ)-改良过的蒙脱石粘土[28]、钪和镧系元素(Ⅲ)盐等。另外,Cu(OTf)2或Sn(OTf)2[29]等也都被成功用作磺酰化催化剂,均可高产率地获得芳香类砜。Olah等[30]发现,Nafion-H也能很好地催化芳烃与磺酸的酰化反应。近几年,一些新型的催化剂如离子液体、离子交换树脂等逐渐出现在人们的视线之中,该类催化剂将会成为人们研究的另一个热点。
2001年,Nara等[31]首次报道了以1-丁基-3-甲基咪唑鎓四氯铝酸盐作为一种非常规的反应介质,在Lewis酸催化下,取代苯与对甲基苯磺酰氯发生Friedel-Crafts反应。反应底物表现出较好的反应活性,几乎定量得到了二苯基砜。与传统的Friedel-Crafts反应相比具有产率高、反应时间短、绿色环保等优点(式(6))。
2001年,Marquie等[32]在超声波辐射下,以FeCl3作为催化剂,磺酰氯或磺酸酐与活泼的芳烃(醚、烷基苯、萘)在110℃下反应1~5 min,以高产率得到了砜类化合物,让人惊喜的是对于较不活泼的芳烃(如苯、苯甲醚、卤代苯等),仅需升高温度和延长反应时间,也可高产率得到砜类化合物。FeCl3与其他金属卤化物尤其是AlCl3相比表现出了更高的催化活性,同时,具有廉价、环境友对等特点。
2004年,Garzya等[33]报道了一种用4-氟苯磺酰氯与取代苯甲醚在InCl3或三氟甲磺酸的催化下快速合成二苯基砜的方法。在对反应底物的适应性研究中他们发现,无论是反应活性较好的卤素取代苯甲醚还是反应活性较差的卤代芳烃,在该反应条件下,均可高产率得到目标化合物,且产物单一。与前面报道的Bi(OTf)3(产率52%)[34]或AlCl3(产率43%)[26]催化下的该类反应相比,具有反应产率高、时间短且立体选择性高的特点。
传统的固体催化剂大多具有不可回收、高腐蚀性、反应加热时间长等缺点。为解决以上问题,研究者将目光集中在新型催化剂的选择上。例如,2004年,Singh等[35]用Fe-PILC作催化剂,以芳烃和磺酰氯为底物,120℃下较短时间内高产率合成了一系列砜类化合物(式(7))。该反应体系表现出优异的催化性能,对反应活性较好的芳烃和活性较差的杂环化合物均适用,且具有反应时间短、产率高、操作简单、催化剂可回收使用等优点。
2009年,Noronha等[36]报道了一类新Friedel-Crafts磺酰化反应,在新型催化剂MoO2Cl2作用下,取代芳烃与对甲苯磺酰氯可以中等到较高产率得到砜类化合物。该反应有效拓展了高价MoO2Cl2化合物在有机合成中的应用,开辟了该类化合物的催化新领域;但不足的是该类反应的反应时间较长,空间位阻及芳环所带的电子对产率的影响较明显,且对于脂肪烃类磺酰氯不适用(图式 4)。
 图式 4
MoO2Cl2催化下Friedel-Crafts取代反应
Scheme4.
MoO2Cl2 as a novel catalyst for Friedel-Crafts sulfonylation
图式 4
MoO2Cl2催化下Friedel-Crafts取代反应
Scheme4.
MoO2Cl2 as a novel catalyst for Friedel-Crafts sulfonylation
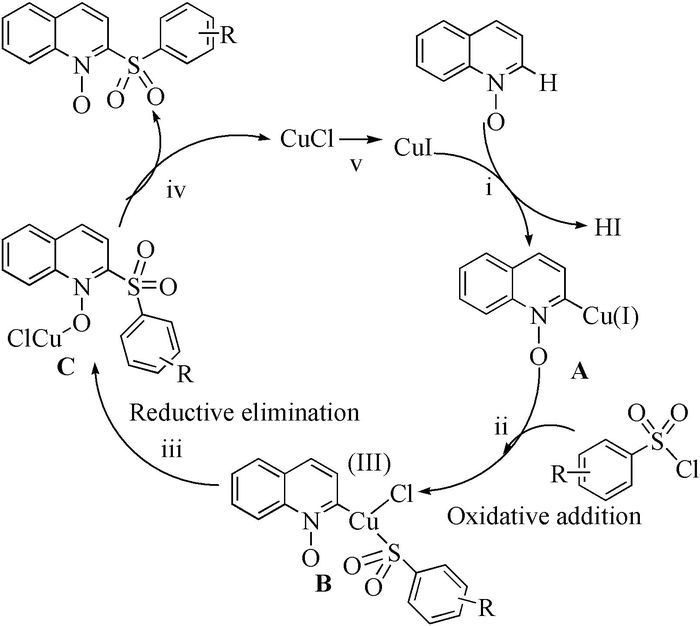
2013年,Wei等[37]报道了一种铜催化的氨基喹啉C-H键与芳基磺酰氯的直接芳基化反应,该反应在氩气保护下进行,在喹啉环的C5-H位上发生取代,但对于脂肪族磺酰氯在该条件下很难得到预期的产物(式(8))。随之,Wu等[38]用CuⅠ为催化剂,以8-氨基喹啉酰胺衍生物和磺酰氯为底物,在空气中100℃下反应24h,也成功实现了8-氨基喹啉C5位C-H键的磺酰化反应(图式 5)。在对反应底物适用范围进行研究的过程中发现,当磺酰氯上带有吸电子基团(F,Cl,Br,CF3,NO2)时所得到的产物产率相对于带有供电子基团(Me,tBu)或电中性时偏低。但令人高兴的是,对于杂环、脂肪族的磺酰氯仍然能以中等产率得到相应的砜类化合物。该反应体系与Wei等的反应体系相比较,其操作更为简单,条件更为温和,同时底物适用范围广。本反应为合成杂环、脂肪族砜类化合物提供了一种新思路。
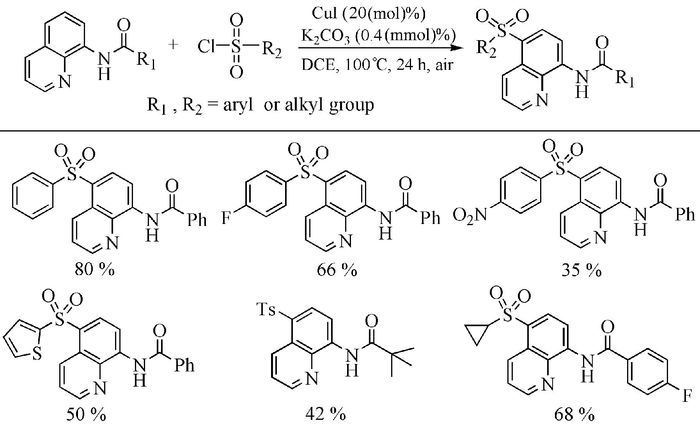 图式 5
CuⅠ催化的8-氨基喹啉酰胺衍生物与磺酰氯的磺酰化反应
Scheme5.
Copper(Ⅰ)-catalyzed sulfonylation of 8-aminoquinoline amides with sulfonyl chlorides
图式 5
CuⅠ催化的8-氨基喹啉酰胺衍生物与磺酰氯的磺酰化反应
Scheme5.
Copper(Ⅰ)-catalyzed sulfonylation of 8-aminoquinoline amides with sulfonyl chlorides
该类反应的反应机理为:首先,CuⅠ作为亲电试剂进攻喹啉N-O邻位碳,得到Cu(Ⅰ)中间体A,接着芳基磺酰氯氧化加成到中间体A上形成Cu(Ⅲ)中间体B,中间体B与Cu原子和N-O原子共同作用生成中间体C,中间体C转化成目标化合物和CuCl,CuCl与I-进行离子交换过程再形成催化剂的循环(图式 6)。
磺酰氯的取代反应大多仅适用于芳基砜的合成,对脂肪族或连有吸电子基团的芳基砜均表现出产率偏低的现象。2017年,Fu等[39]报道了一种金属镁介导的超声波辐射下一锅法、两步合成脂肪族砜的方法。他们发现,在超声波辐射下,有机磺酰氯与Mg在THF中可高产率地转化成亚磺酸镁盐,在亚磺酸镁盐中加入催化量的NaI,并以DMSO作为助溶剂(THF:DMSO=3:1),60℃时反应1h,较高产率地得到了一系列脂肪族砜(式(9))。该方法具有操作简单、反应时间短、原料廉价等优点,值得一提的是,他们首次完成了由磺酰氯到无水亚磺酸盐的直接转化,为无水亚磺酸盐的合成及脂肪族砜类化合物的大规模生产提供了新选择。
3 亚磺酸盐的偶联反应
近年来,芳基亚磺酸盐与芳基卤化物或芳基硼酸的交叉偶联反应作为一种非常规合成砜类化合物方法备受人们关注。根据催化剂的使用情况,该类反应主要有以下三种。
3.1 过渡金属催化的偶联反应
自1983年,钯作为催化剂首次催化完成了溴苯的胺化反应后[40],过渡金属作为催化剂也被人们尝试着应用于芳基卤化物、有机硼酸等底物的磺酰化反应,并取得了突破性的进展。Cacchi等[41]报道了一种Pd2(dba)3催化的芳基和乙烯基卤化物或三氟甲磺酸酯与亚磺酸钠的偶联反应,该反应以Cs2CO3为碱,在钯直接催化下首次将Caryl-X(X=I,Br,OTf)转化为Caryl-SO2,其中nBu4NCl和膦配体对反应起着重要作用(式(10))。
1995年,Suzuki等[42]首次报道了芳基卤化物与亚磺酸钠在CuⅠ催化下,以较高产率得到不对称的二苯基砜类化合物,自此,人们将研究的目光转移到了铜催化的偶联反应。2002年,Baskin等[43]报道了一个更高效的催化过程,以((CuOTf)2PhH)为催化剂、N, N’-二甲基乙二胺为配体,110℃下反应20h,可以24%~96%的产率得到二苯基砜。不足之处是该反应要求的温度较高、反应时间较长,且底物只能限定在反应活性较好的碘代芳烃,以活性较低的溴代芳烃和杂环芳烃为底物时产率偏低。
2004年,Beaulieu等[44]报道了芳基硼酸与亚磺酸钠盐在过量Cu(OAc)2催化及K2CO3和分子筛存在的条件下,室温于DMSO中发生交叉偶联反应,高产率地得到一系列带各种官能团的砜类化合物。但不足的是该反应需要过量的催化剂,不适合工业化生产,且底物为杂环取代的硼酸时,反应效果不太理想(式(11))。
2005年,Zhu等[45]发现一些氨基酸能促进CuⅠ催化C-N、C-C、C-O键的形成。基于此,他们探索了卤代芳烃与亚磺酸盐用CuⅠ/L-脯氨酸钠作为催化体系的偶联反应,发现底物在DMSO中80~90℃下很容易发生偶联反应,可高产率得到芳基砜。该类反应具有很好的兼容性,当卤代芳烃上连有-OH、-NH3、-OCH3、-CN、-COCH3等官能团时均不受影响。并且,不活泼的溴代芳烃和溴代杂环在该反应条件下均能反应得到相应的砜,而当溴代芳烃上连有富电子基团比连有缺电子基团时反应活性更高(式(12))。可能是较低的反应温度更适合该反应体系的进行,而确切的反应机理还有待进一步研究。
Kuwano等[46]研究发现,用苄基碳酸盐(以相应的醇为原料,与氟甲酸甲酯发生酯化反应快速得到)与芳烃类亚磺酸盐在钯复合体[Pd(η3-C3H5)Cl]2-DPEphos存在的条件下,也可发生反应生成砜类化合物(式(13)),并对反应机理进行了介绍。该催化反应体系为苄基砜的合成提供了新的途径,拓展了合成砜的底物范围。
配体可加速芳基硼酸与亚磺酸盐的交叉偶联反应。2007年,Kar等[47]和Huang等[48]先后报道了在不加碱的情况下,配体1, 10-菲咯啉和1-苄基咪唑(式(14))的使用可大大减少铜盐的用量,在分子筛和催化量Cu(OAc)2条件下,均可以较高产率得到甲基-芳基、二芳基和杂环芳基及烯基砜类化合物(图式 7)。该方法有效地将底物范围拓展到了烯基和杂环硼酸,且大大减少了催化剂用量,避免了碱的使用。但缺点是这些方法都需要较长的反应时间,还需加入配体。
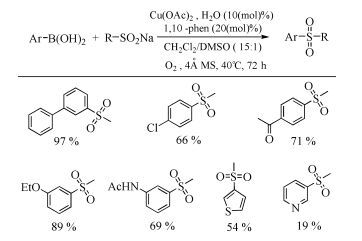 图式 7
Cu(OAc)2催化的硼酸与亚磺酸盐的交叉偶联反应
Scheme7.
Cu(OAc)2 catalyzed cross-coupling reaction of boronic acids and sulfinic acid salts
图式 7
Cu(OAc)2催化的硼酸与亚磺酸盐的交叉偶联反应
Scheme7.
Cu(OAc)2 catalyzed cross-coupling reaction of boronic acids and sulfinic acid salts
离子液体被人们公认为是挥发性有机溶剂的替代品,具有无味、无恶臭、无污染、易与产物分离、易回收、可反复多次循环使用等优点,因而备受人们青睐。2008年,Kantam等[49]报道了绿色合成烷基芳基、二芳基砜的合成方法,使用催化量的Cu(OAc)2在离子液体作用下,较短时间内、无配体条件下芳基硼酸与亚磺酸盐发生偶联反应,该反应具有操作简单、反应条件温和、催化剂可循环使用、对环境友好等优点(式(15))。
2011年,Zhou等[50]在Pd作催化剂的Heck脱硫偶联反应中发现存在着脱硫反应和共轭加成反应的竞争。以苯甲醚作溶剂、DPEphos作配体时,可高产率(95%)得到脱硫反应的产物;用DMSO作溶剂、L-苯丙氨酸作配体时,可较高产率(60%)地得到共轭加成反应的产物砜(式(16))。
2012年,Wu等[51]首次报道了一种高效的烯丙基胺与亚磺酸盐的偶联反应。在0.1(mol)% [Pd(allyl)Cl]2、0.4(mol)% 1, 4-双(二苯膦基)丁烷(dppb)和过量的硼酸存在条件下,以1, 4-二氧六环为溶剂,100℃下反应4h,α-支链的烯丙基伯胺很容易被亚磺酸钠取代,高产率(67%~94%)地得到了烯丙基砜。为了进一步研究手性烯丙基砜的合成,他们用外消旋1, 1′-联-2-萘酚(BINOL)作配体代替dppb,用一个光学纯度为99% ee的α-手性烯丙基伯胺作底物,在上述其他条件不变的情况下得到了手性烯丙基砜,产率97%,96% ee。该反应从不对称的烯丙基亲电试剂着手,为合成具有高光学纯度的α-手性烯丙基砜提供了一种简单可行的方法(式(17))。
2013年,Cullen等[52]首次报道了一种用铜催化二芳基碘盐与廉价易得的三氟甲基磺酸盐合成三氟甲基砜的方法。该方法与其他钯或铜催化的偶联反应相比较,无需配体或碱性助剂,成功地在芳环上引入三氟甲基砜基,同时,该类反应对芳基上的氯、溴、酯和硝基等基团都有较好的耐受力,且杂环碘代盐也可在该条件下得到较满意的产率(图式 8)。
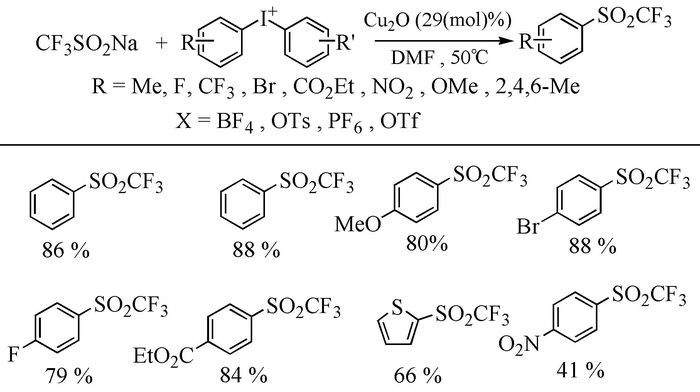 图式 8
Cu催化的芳基碘盐与三氟甲基磺酸钠的偶联反应
Scheme8.
Cu-catalyzed couplings of aryliodonium salts with sodium trifluoromethanesulfinate
图式 8
Cu催化的芳基碘盐与三氟甲基磺酸钠的偶联反应
Scheme8.
Cu-catalyzed couplings of aryliodonium salts with sodium trifluoromethanesulfinate
2014年,Tang等以Cu(OAc)2作催化剂、亚磺酸钠与肟乙酸酯为底物,在N2保护下,100℃反应6h,高产率合成了一系列砜的衍生物[53](图式 9)。该反应最大的优点在于利用了肟乙酸酯自身氧化性能,使其既作反应底物又作氧化剂,有效避免了外部氧化剂和催化剂的使用,为氧化偶联合成砜开辟了新的路线和方向。另外,在对底物适应性的探讨时发现,当肟乙酸酯被其他官能团取代时,吸电子基团对反应没有表现出明显的影响,但遗憾的是对烷基肟乙酸酯不适合该反应。对于取代的亚磺酸钠,供电子基团可有利于产率的提高,同时无论是芳香类还是脂肪类、脂环类的亚磺酸钠,均可在该条件下获得相应的砜的衍生物。
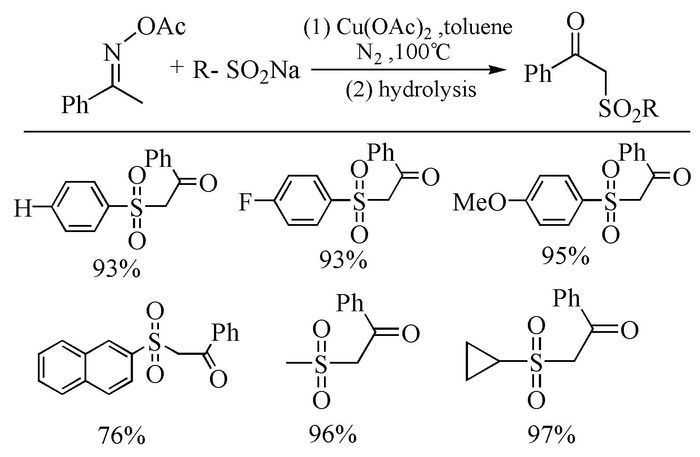 图式 9
铜催化的亚磺酸钠与肟乙酸酯的氧化偶联反应
Scheme9.
Copper-catalyzed oxidative coupling reaction of oxime acetates with sodium sulfinates
图式 9
铜催化的亚磺酸钠与肟乙酸酯的氧化偶联反应
Scheme9.
Copper-catalyzed oxidative coupling reaction of oxime acetates with sodium sulfinates
过渡金属催化的偶联反应,尽管选择性和产品收率上具有显著优势,且避免了取代反应自身固有的一些缺陷,但该类反应也具有潜在的金属污染,不适合大规模的工业化生产,尤其是对于金属污染检测严格的制药工业。而且,反应底物为卤代芳烃时,仅限于溴代烃、碘代烃和三氟甲磺酸酯,而这些物质均表现出对空气和水较敏感的特性,相对来说,反应条件较苛刻,同时,所用催化剂高昂的价格、较强的毒性以及对含磷配体的依赖严重制约了它在很多领域的工业化应用。
3.2 非金属催化的偶联反应
I2作为一种温和、廉价易得的固体催化剂,与溴、氯相比操作容易,成为氧化偶联反应的首选催化剂,吸引了广大化学工作者的注意。2014年,Xiao等[54]用I2作催化剂、过氧化叔丁醇(TBHP)为氧化剂、乙酸作溶剂,取代吲哚与亚磺酸盐室温反应1~4 h,高产率得到2-位取代芳基砜基吲哚。在对反应区域选择性的研究中他们发现,当吲哚环的4-C和7-C带有-CH3时,2-C的取代产物产率可达89%~90%;当5-X取代时,产率会下降34%~50%;当2-C上连有其他官能团时,只在3-C上获得微量的砜基吲哚。由此可见,该类反应具有极高的区域选择性。同时,在对底物范围进行研究时,以取代的亚磺酸盐为研究对象,发现无论是芳烃类还是脂肪族的亚磺酸盐,该方法均适用。该方法提供了一种简单、有效制备杂环类化合物的方法,且其高区位选择性使吲哚的2-C位上的官能团化有了新的突破。一般情况下,3-C位要比2-C位优先取代,但由于-NH影响,使2-C位被活化,从而高产率得到2-位取代的砜类化合物(图式 10)。
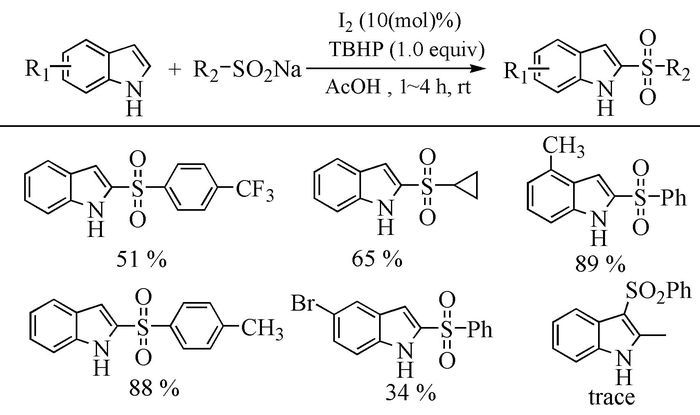 图式 10
I2催化亚磺酸盐和吲哚的区域选择性2-磺酰化反应
Scheme10.
Iodine-catalyzed regioselective 2-sulfonylation of indoles with sodium sulfinates
图式 10
I2催化亚磺酸盐和吲哚的区域选择性2-磺酰化反应
Scheme10.
Iodine-catalyzed regioselective 2-sulfonylation of indoles with sodium sulfinates
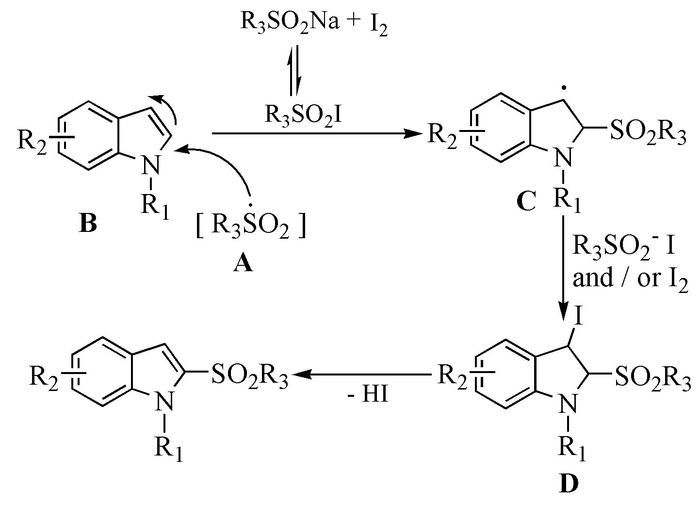
随后,Katrun等[55]也报道了I2介导的吲哚2-C上的磺酰化反应,该反应无需氧化剂,于甲醇中室温反应2h即可高产率地得到2-位取代砜基吲哚的衍生物。与前一个反应相比较,他们对试剂的用量、溶剂、物料顺序、反应温度、光照条件和底物范围进行了更为详细的研究,发现底物的物料比、溶剂、物料加入顺序对产物产率的影响比较明显,而反应温度和光照条件等的影响不明显,并根据反应结果对反应机理进行了推测。该类反应据信是经过以下反应历程(图式 11):亚磺酸盐与I2反应得到磺酰基碘的中间体A,A发生均裂,产生磺酰基自由基B,B与吲哚发生加成反应生成吲哚自由基C,C结合来自于I2单质或磺酰基碘中的碘生成D,D发生消除反应脱去HI分子,得到预期的目标化合物砜基吲哚。
2015年,Xiao等[56]报道了一种简便、高效的制备2-砜基甲基喹啉的方法。以2-甲基喹啉衍生物与亚磺酸盐为反应物,在KI作催化剂、TBHP为氧化剂条件下,于空气中80℃反应16h,高产率得到了一系列杂环苄基砜(式(18))。在该反应体系中,当芳基亚磺酸钠的芳基连有-X、-CF3、-OCF3等官能团时,仍然能以较高收率(62%~72%)得到相应的产物。经GC-MS检测,未发现C-X键断裂;同时,除了芳基亚磺酸盐外,活性较差的脂肪族亚磺酸盐也适用于此反应。
2015年,Chen等[57]以肉桂酸与苯亚磺酸钠为底物,在I2和TBHP催化下,在甲苯中90℃反应12h,通过脱羧、偶联高产率地合成烯基砜(式(19))。该方法反应底物肉桂酸廉价易得,且避免了金属催化剂的使用,为烯基砜的合成提供了新的思路。
随后,Yadav等[58]也报道了一种I2诱导下的氧化偶联制备β-酮砜的反应。用烯醇乙酸酯和亚磺酸钠为底物,无需任何配体,于CH3CN/H2O(4:1,体积比)混合溶剂中,90℃下反应10~12 h,以较高产率得到了一系列的β-酮砜(式(20))。该方法有效避免了过渡金属和配体的使用,为β-酮砜的合成提供了一种实用、经济、安全、温和、对环境友好的方法。在对反应条件进行探索的过程中发现,反应底物无论连有吸电子基团还是供电子基团对产率的影响均不明显。
3.3 无催化剂的偶联反应
2011年,Maloney等[59]用一种活泼的卤代吡啶与亚磺酸钠盐为底物,无催化剂和配体,一锅法合成含吡啶的砜类化合物。他们发现,当卤代吡啶连有缺电子基团时,反应中加入四丁基氯化铵(TBACl),可高产率地得到含吡啶的砜类化合物。当卤代吡啶呈电中性或连有富电子基团时,在以上条件下,反应几乎不能发生;但当在反应体系中加入等摩尔的浓HCl时,同样可以高产率得到含吡啶的砜类化合物。TBACl与NaSO2Tol作用形成n-Bu4NSO2Tol,而n-Bu4NSO2Tol在DMAc中的溶解度要大于NaSO2Tol,进而促进反应的顺利进行。该方法不仅适用于含吡啶环的卤代烃,同时还适用于含其他杂环如吡嗪、吲哚等的卤代烃,反应具有条件温和、底物来源方便、廉价,操作简单、安全、绿色,且空间位阻对产率影响不是很大等优点(图式 12)。
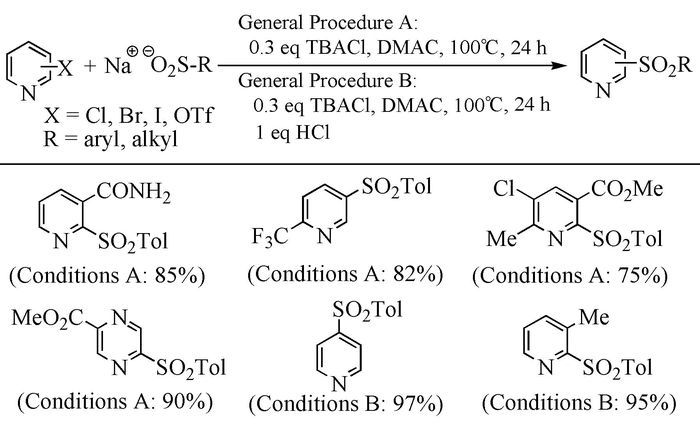 图式 12
卤素取代吡啶与亚磺酸盐的反应
Scheme12.
Synthesis of sulfonylated pyridines via the direct SNAr displacement of halogen substituted pyridines with sodium sulfinate salts
图式 12
卤素取代吡啶与亚磺酸盐的反应
Scheme12.
Synthesis of sulfonylated pyridines via the direct SNAr displacement of halogen substituted pyridines with sodium sulfinate salts
2013年,Liang等[60]用含有一个或多个杂原子的卤代或硝基取代五元杂环与亚磺酸盐反应以较高产率得到含五元杂环砜类化合物(式(21))。在过渡金属催化的反应中,氯代噻吩比溴代噻吩的产率低,而该反应则刚好解决了这一问题,在该反应条件下,可使氯代噻吩与溴代噻吩的产率几乎完全相同。
2013年,Manolikakes等[61]利用二芳基碘盐强亲电试剂的特性代替芳基化试剂来作为硫的亲核试剂,在无任何金属催化和碱性助剂的条件下,在非质子溶剂如DMF、DMSO、NMP中,90℃下反应24h,高产率得到二苯基砜。该反应对水和空气不敏感,无需惰性气体保护,溶剂也不用做无水处理,且空间位阻对产率影响不是很大,反应底物无论是芳烃类还是杂环类的亚磺酸盐均适用,有效解决了杂环取代和空间位阻大的问题(式(22))。
苯炔是一类重要的具有高度反应活性的有机合成中间体[62~67],但由于其活性较高而不易分离获得,故都是以合成其前体加以代替。Pandya等[68]发现烷基、芳基或杂环类取代的亚磺酸钠与不同取代的苯炔前体在无需任何催化剂的条件下,以四丁基氟化铵(TBAF)作为氟源和相转移催化剂,室温下反应可高产率地得到多种类型的芳基砜。该方法无需加热在室温即可较高产率的得到目标化合物,方法简便、反应速度快(式(23))。
4 结语
本文综述了近年来砜类化合物合成方法的研究进展,从上述的讨论可以看出,该领域多年来已经引起了人们广泛关注并取得了较大的进展。硫醚的氧化反应底物大多都来自于带有难闻气味的硫醇,且一些氧化剂在反应过程中产生等量的低价态还原产物,会增加产物分离提纯的难度,处理这些废弃物时,会对环境产生不良影响。这些缺点使其在工业上的应用受到了一定的限制。用Lewis酸或质子酸作催化剂的取代反应,虽然具有原料廉价易得、反应条件温和、对环境较友好的特点,但该类反应多受空间位阻与取代基电子效应的影响,对产物的邻、对位同分异构体具有较低的选择性。过渡金属催化的亚磺酸盐与芳卤代烃、有机硼酸等偶联反应近年来也得到了迅速的发展,研究者们围绕反应底物的拓展、高效催化剂选择性的提高等方面开展了大量工作。但是这些偶联反应也具有一定的缺点,如过渡金属价格昂贵、对环境有一定的污染、底物适应范围窄、反应条件苛刻、亚磺酸盐较难获得等。虽然在无金属催化、无需配体和碱性助剂的条件下合成砜类化合物有望改善这一状况,但是此类反应唯一不足的是反应时间较长,反应底物多限于卤代烃或类卤化物,故能寻找到一种高效、廉价、对环境无污染的催化剂或多种能代替亚磺酸盐的亲核试剂也是该类反应今后研究和发展的方向。
-
-
[1]
B J Zhang, A M Wassermann, M Vogt et al. J. Chem. Inf. Model., 2012, 52(12):3138~3143. doi: 10.1021/ci300481d
-
[2]
J S Scott, A M Birch, K J Brocklehurst et al. J. Med. Chem., 2012, 55:5361~5379. doi: 10.1021/jm300310c
-
[3]
W M Xu, F F Han, M He et al. J. Agric. Food Chem., 2012, 60:1036~1041. doi: 10.1021/jf203772d
-
[4]
M C Carreno. Chem. Rev., 1995, 95:1717~1726. doi: 10.1021/cr00038a002
-
[5]
M Artico, R Silvestri, S Massa et al. J. Med. Chem., 1996, 39(2):522~530. doi: 10.1021/jm950568w
-
[6]
T M Williams, T M Ciccarone, S C Mac Tough et al. J. Med. Chem., 1993, 36(9):1291~1294. doi: 10.1021/jm00061a022
-
[7]
W T Li, D R Hwang, J S Song et al. J. Med. Chem., 2010, 53:2409~2417. doi: 10.1021/jm901501s
-
[8]
R Silvestri, M Artico, G L Regina et al. Farmaco, 2004, 59:201~210. doi: 10.1016/j.farmac.2003.11.004
-
[9]
A Basak, M Goswami, A Rajkumar et al. Bioorg. Med. Chem. Lett., 2015, 25(10):2225~2237.
-
[10]
S Consalvia, S Alfonsoa, A D Capuab et al. Bioorg. Med. Chem. Lett., 2015, 23(4):810~820.
-
[11]
L M Ni, X S Zheng, P K Somers et al. Bioorg. Med. Chem. Lett., 2003, 13(4):745~748.
-
[12]
D H Boschelli, J B Kramer, S S Khatana et al. J. Med. Chem., 1995, 38(22):4597~4614. doi: 10.1021/jm00022a026
-
[13]
T K Pal, S Dey, T Pathak. J. Org. Chem., 2011, 76(9):3034~3041. doi: 10.1021/jo101877r
-
[14]
D Lee, C L Williamson, L N Chan et al. J. Am. Chem. Soc., 2012, 134:8260~8267. doi: 10.1021/ja302549c
-
[15]
R Aldea, H Alper. J. Org. Chem., 1995, 60:8365~8366. doi: 10.1021/jo00131a009
-
[16]
R Beckerbauer, B E Smart. J. Org. Chem., 1996, 60:6186~6187.
-
[17]
R S Varma, R K Saini, H M Meshram. Tetrahed. Lett., 1997, 38:6525~6528. doi: 10.1016/S0040-4039(97)01520-7
-
[18]
L Xu, J Cheng, M L Trudell. J. Org. Chem., 2003, 68:5388~5391. doi: 10.1021/jo030031n
-
[19]
刘课艳, 偶辉, 石先莹等.有机化学, 2014, 34, 681~692. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=yjhu201404005&dbname=CJFD&dbcode=CJFQ
-
[20]
K Bahrami. Tetrahed. Lett., 2006, 47:2009~2012. doi: 10.1016/j.tetlet.2006.01.051
-
[21]
M Rahimizadeh, G Rajabzadeh, S M Khatami et al. J. Mol. Catal. A, 2010, 323:59~64. doi: 10.1016/j.molcata.2010.03.011
-
[22]
A Rostami, J Akradi. Tetrahed. Lett., 2010, 51:3501~3503. doi: 10.1016/j.tetlet.2010.04.103
-
[23]
M Jereb. Green Chem., 2012, 14:3047~3052. doi: 10.1039/c2gc36073j
-
[24]
B Maleki, S Hemmati, A Sedrpoushan et al. RSC Adv., 2014, 4:40505~40510. doi: 10.1039/C4RA06132B
-
[25]
R D Chakravarthy, V Ramkumara, D K Chand. Green Chem., 2014, 16:2190~2196. doi: 10.1039/c3gc42245c
-
[26]
B Graybill. J. Org. Chem., 1967, 32(9):2931~2933. doi: 10.1021/jo01284a075
-
[27]
C G Frost, J P Hartleya, D Griffinb. Tetrahed. Lett., 2002, 43:4789~4791 doi: 10.1016/S0040-4039(02)00931-0
-
[28]
G A Olah, A Orlinkov, A B Oxyglou et al. J. Org. Chem., 1985, 50(8):1306~1309. doi: 10.1021/jo00208a034
-
[29]
R P Singh, R M Kamble, K L Chandra et al. Tetrahed. Lett., 2001, 57:241~247. doi: 10.1016/S0040-4020(00)01005-X
-
[30]
G A Olah, T Mathew, G K S Parakash. Chem. Commun., 2001, 1696~1697.
-
[31]
S J Nara, J R Harjani, M F Salunkhe. J. Org. Chem., 2001, 66:8616~8620. doi: 10.1021/jo016126b
-
[32]
J Marquie, A Laporterie, J Dubac. J. Org. Chem., 2001, 66:421~425. doi: 10.1021/jo0010173
-
[33]
V Garzya, I T Forbes, S Lauru et al. Tetrahed. Lett., 2004, 45:1499~1501. doi: 10.1016/j.tetlet.2003.12.028
-
[34]
S Repichet, C Le Roux, P Hernandez et al. J. Org. Chem., 1999, 64(17):6479~6483. doi: 10.1021/jo9902603
-
[35]
D U Singh, P R Singh, S D Samant. Tetrahed. Lett., 2004, 45:9079~9082. doi: 10.1016/j.tetlet.2004.10.039
-
[36]
R G D Noronha, A C Fernandes, C C Romão. Tetrahed. Lett., 2009, 50:1407~1410. doi: 10.1016/j.tetlet.2009.01.039
-
[37]
H W Liang, K Jiang, W Ding et al. Chem. Commun., 2015, 51:16928~16931. doi: 10.1039/C5CC05527J
-
[38]
Z Y Wu, H Y Song, X L Cui et al. Org. Lett., 2013, 15(6):1270~1273. doi: 10.1021/ol400178k
-
[39]
Y Fu, Q S Xu, Q Z Li et al. Org. Biomol. Chem., 2017, 13:2841~2845.
-
[40]
刘蒲, 李三华, 李利民等.化学进展, 2005, 17(2):281~291.
-
[41]
S Cacchi, G Fabrizi, A Goggiamani et al. J. Org. Chem., 2004, 69:5608~5614. doi: 10.1021/jo0493469
-
[42]
H Suzuki, H Abe. Tetrahed. Lett., 1995, 36:6239~6242. doi: 10.1016/0040-4039(95)01095-Y
-
[43]
J M Baskin, Z Wang. Org. Lett., 2002, 4(25):4423~4425. doi: 10.1021/ol0269190
-
[44]
C Beaulieu, D Guay, Z Y Wang et al. Tetrahed. Lett., 2004, 45:3233~3236. doi: 10.1016/j.tetlet.2004.02.127
-
[45]
W Zhu, D W Ma. J. Org. Chem., 2005, 70:2696~2700. doi: 10.1021/jo047758b
-
[46]
R Kuwano, Y Kondo, T Shirahama. Org. Lett., 2005, 7(14):2973~2975. doi: 10.1021/ol0509787
-
[47]
A Kar, I A Sayyed, W F Lo et al. Org. Lett., 2007, 9(17):3405~3408. doi: 10.1021/ol071396n
-
[48]
F Huang, R A Batey. Tetrahedron, 2007, 63:7667~7672. doi: 10.1016/j.tet.2007.05.029
-
[49]
M L Kantam, B Neelima, B Sreedhar et al. Synlett, 2008, 10:1455~1458.
-
[50]
X Y Zhou, J Y Luo, Liu J et al. Org. Lett., 2011, 13(6):1432~1435. doi: 10.1021/ol200101x
-
[51]
X S Wu, Y Chen, M B Li et al. J. Am. Chem. Soc., 2012, 134:14694~14698. doi: 10.1021/ja306407x
-
[52]
S C Cullen, S Shekhar, N K Nere. J. Org. Chem., 2013, 78:12194~12201. doi: 10.1021/jo401868x
-
[53]
X D Tang, L B Huang, Y L Xu et al. Angew. Chem. Int. Ed., 2014, 53:4205~4208.
-
[54]
F H Xiao, H Chen, H Xie et al. Org. Lett., 2014, 16:50~53. doi: 10.1021/ol402987u
-
[55]
P Katrun, C Mueangkaew, M Pohmakotr et al. J. Org. Chem., 2014, 79(4):1778~1785. doi: 10.1021/jo402831k
-
[56]
F H Xiao, S Q Chen, Y Chen et al. Chem. Commun., 2015, 51:652~654. doi: 10.1039/C4CC07546C
-
[57]
J Chen, J C Mao, Y Zheng et al. Tetrahedron, 2015, 71(31):5059~5063. doi: 10.1016/j.tet.2015.05.115
-
[58]
V K Yadav, V P Srivastava, L D S Yadav. Tetrahed. Lett., 2016, 57(21):2236~2238. doi: 10.1016/j.tetlet.2016.04.018
-
[59]
K M Maloney, J T Kuethe, K Linn. Org. Lett., 2011, 13(1):102~105. doi: 10.1021/ol102629c
-
[60]
S Liang, R Y Zhang, L Y Xi et al. J. Org. Chem., 2013, 78:11874~11880. doi: 10.1021/jo401828b
-
[61]
N Umierski, G Manolikakes. Org. Lett., 2013, 15(1):188~191. doi: 10.1021/ol303248h
-
[62]
李连贵.松辽学报(自然科学版), 2001, 2:40~43.
-
[63]
Z J Liu, R C Larock. Org. Lett., 2004, 6:99~102. doi: 10.1021/ol0361406
-
[64]
Z J Liu, R C Larock. J. Am. Chem. Soc., 2005, 127(38):13112~13113. doi: 10.1021/ja054079p
-
[65]
U K Tambar, B M Stoltz. J. Am. Chem. Soc., 2005, 127(15):5340~5341. doi: 10.1021/ja050859m
-
[66]
T T Tayan, M Jeganmohan, M J Cheng. J. Am. Chem. Soc., 2006, 128(7):2232~2233. doi: 10.1021/ja058418q
-
[67]
J L Henderson, A S Edwards, M F Greaney. J. Am. Chem. Soc., 2006, 128(23):7426~7427. doi: 10.1021/ja0615526
-
[68]
V G Pandya, S B Mhaske. Org. Lett., 2014, 16:3836~3839. doi: 10.1021/ol5018646
-
[1]
-

图式 1 含有芳基砜的代表性药物和农药
Scheme 1 Typical drugs and pesticide containing aryl sulfone motif

图式 2 H5IO6/CrO3选择性将硫醚氧化成砜
Scheme 2 H5IO6/CrO3 chemoselective oxidation of sulfides to sulfones

图式 3 无溶剂条件下用H2O2和硼酸作催化剂的硫醚氧化反应
Scheme 3 A oxidation of sulfides using hydrogen peroxide and boric acid as the catalyst under solvent-free conditions

图式 4 MoO2Cl2催化下Friedel-Crafts取代反应
Scheme 4 MoO2Cl2 as a novel catalyst for Friedel-Crafts sulfonylation

图式 5 CuⅠ催化的8-氨基喹啉酰胺衍生物与磺酰氯的磺酰化反应
Scheme 5 Copper(Ⅰ)-catalyzed sulfonylation of 8-aminoquinoline amides with sulfonyl chlorides

图式 7 Cu(OAc)2催化的硼酸与亚磺酸盐的交叉偶联反应
Scheme 7 Cu(OAc)2 catalyzed cross-coupling reaction of boronic acids and sulfinic acid salts

图式 8 Cu催化的芳基碘盐与三氟甲基磺酸钠的偶联反应
Scheme 8 Cu-catalyzed couplings of aryliodonium salts with sodium trifluoromethanesulfinate

图式 9 铜催化的亚磺酸钠与肟乙酸酯的氧化偶联反应
Scheme 9 Copper-catalyzed oxidative coupling reaction of oxime acetates with sodium sulfinates

图式 10 I2催化亚磺酸盐和吲哚的区域选择性2-磺酰化反应
Scheme 10 Iodine-catalyzed regioselective 2-sulfonylation of indoles with sodium sulfinates
-


 下载:
下载:











 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 0
- HTML全文浏览量: 0
 下载:
下载:
