-
[1]
WANG A Y, WANG Y L, WALTER E D, KUKKADAPU R K, GUO Y L, LU G Z, WEBER R S, WANG Y, PEDEN C H F, GAO F. Catalytic N2O decomposition and reduction by NH3 over Fe/Beta and Fe/SSZ-13 catalysts[J]. J Catal,
2018, 358:
199-210.
doi: 10.1016/j.jcat.2017.12.011
-
[2]
徐向阳, 谷成, 王虹, 张远远, 柯琰, 张成乐, 王明锦, 宋宝华, 李翠清. Co/Hβ催化剂上N2O的分解性能研究[J]. 燃料化学学报,
2014,42,(7): 877-883.
XU Xiang-yang, GU Cheng, WANG Hong, ZHANG Yuan-yuan, KE Yan, ZHANG Cheng-le, WANG Ming-jin, SONG Bao-hua, LI Cui-qing. Catalytic performance of Co/Hβ in N2O decomposition[J]. J Fuel Chem Technol,
2014, 42(7):
877-883.
-
[3]
王虹, 王军利, 李翠清, 宋永吉, 迟姚玲, 王焘. ACo2O4/HZSM-5催化剂上N2O的直接分解[J]. 物理化学学报,
2010,26,(10): 2739-2744.
doi: 10.3866/PKU.WHXB20100928WANG Hong, WANG Jun-li, LI Cui-qing, SONG Yong-ji, CHI Yao-ling, WANG Tao. Decomposition of N2O on ACo2O4/HZSM-5 Catalysts[J]. Acta Phys Chim Sin,
2010, 26(10):
2739-2744.
doi: 10.3866/PKU.WHXB20100928
-
[4]
LIU Z M, HE F, MA L L, PENG S. Recent advances in catalytic decomposition of N2O on noble metal and metal oxide catalysts[J]. Catal Surv Asia,
2016, 20(3):
1-12.
-
[5]
KONSOLAKIS M. Recent advances on nitrous oxide (N2O) decomposition over non-noble metal oxide catalysts:Catalytic performance, mechanistic considerations and surface chemistry aspects[J]. Acs Catal,
2015, 5:
6397-6421.
doi: 10.1021/acscatal.5b01605
-
[6]
YAKOVLEV A L, ZHIDOMIROV G M, VAN SANTEN R A V. N2O decomposition catalyzed by transition metal ions[J]. Catal Lett,
2001, 75:
45-48.
doi: 10.1023/A:1016692419859
-
[7]
FELLAH M F, ONAL I. N2O decomposition on Fe-and Co-ZSM-5:A density functional study[J]. Catal Today,
2008, 137:
410-417.
doi: 10.1016/j.cattod.2007.10.114
-
[8]
RYDER J A, CHAKRABORTY A K, BELL A T. Density functional theory study of nitrous oxide decomposition over Fe-and Co-ZSM-5[J]. J Phys Chem B,
2002, 106:
7059-7064.
doi: 10.1021/jp014705e
-
[9]
SUI C, ZHANG T R, DONG Y L, YUAN F L, NIU X Y, ZHU Y J. Interaction between Ru and Co3O4 for promoted catalytic decomposition of N2O over the Rux-Co3O4 catalysts[J]. Mol Catal,
2017, 435:
174-181.
doi: 10.1016/j.mcat.2017.03.033
-
[10]
CHENG H K, HUANG Y Q, WANG A Q, LI L, WANG X D, ZHANG T. N2O decomposition over K-promoted Co-Al catalysts prepared from hydrotalcite-like precursors[J]. Appl Catal B:Environ,
2009, 89(3):
391-397.
-
[11]
WANG Y Z, HU X B, ZHENG K, ZHANG H X, ZHAO Y X. Effect of precipitants on the catalytic activity of Co-Ce composite oxide for N2O catalytic decomposition[J]. React Kinet Mech Catal,
2018, 123(2):
707-721.
doi: 10.1007/s11144-017-1293-9
-
[12]
IVANOVA Y A, SUTORMINA E F, ISUPOVA I A, VOVK E I. Catalytic activity of the oxide catalysts based on Ni0.75Co2.25O4 modified with cesium cations in a reaction of N2O decomposition[J]. Kinet Catal,
2017, 58(6):
793-799.
-
[13]
WANG Y Z, HU X B, ZHENG K, WEI X H, ZHAO Y X. Effect of SnO2 on the structure and catalytic performance of Co3O4 for N2O decomposition[J]. Catal Commun,
2018, 111:
70-74.
doi: 10.1016/j.catcom.2018.04.004
-
[14]
LIU N, CHEN P, LI Y X, ZHANG R D. N2O Direct dissociation over MgxCeyCo1-x-yCo2O4 composite spinel metal oxide[J]. Catalysts,
2017, 7(1):
1-12.
-
[15]
WANG Y Z, HU X B, ZHENG K, ZHANG H X, ZHAO Y X. Effect of precipitants on the catalytic activity of Co-Ce composite oxide for N2O catalytic decomposition[J]. React Kinet Mech Catal,
2018, 123(2):
707-721.
doi: 10.1007/s11144-017-1293-9
-
[16]
DUAN Y K, ZHANG Q L, SONG Z X, WANG J, TANG X S, LIU Q X, ZANG T F. Effect of preparation methods on the catalytic activity of Co3O4 for the decomposition of N2O[J]. Res Chem Intermed,
2017, 43(12):
7241-7255.
doi: 10.1007/s11164-017-3071-8
-
[17]
CIURA K, GRZYBEK G, WOJCIK S, INDYK P, KOTARBA A, SOJKA Z. Optimization of cesium and potassium promoterloading in alkali-doped Zn0.4Co2.6O4 vertical bar Al2O3 catalysts for N2O abatement[J]. React Kinet Mech Catal,
2017, 121(2):
645-655.
doi: 10.1007/s11144-017-1188-9
-
[18]
DOU Z, ZHANG H, PAN Y, XU X F. Catalytic decomposition of NO over potassium-modified Cu-Co spinel oxides[J]. J Fuel Chem Technol,
2014, 42(2):
238-245.
doi: 10.1016/S1872-5813(14)60016-5
-
[19]
ZHU Z Z, LU G Z, ZHANG Z G, GUO Y, GUO Y L, WANG Y Q. Highly active and stable Co3O4/ZSM-5 catalyst for propane oxidation:Effect of the preparation method[J]. Acs Catal,
2013, 3(3):
1154-1164.
-
[20]
ABDALLAH H M I, MOYO T. Structural and magnetic studies of (Mg, Sr)0.2Mn0.1Co0.7Fe2O4 nanoferrites[J]. J Alloy Compd,
2013, 562(11):
156-163.
-
[21]
LOGANATHAN A, KUMAR K. Effects on structural, optical, and magnetic properties of pure and Sr-substituted MgFe2O4 nanoparticles at different calcination temperatures[J]. Appl Nanosci,
2016, 6(5):
629-639.
doi: 10.1007/s13204-015-0480-0
-
[22]
刘畅, 薛莉, 贺泓. 碱土金属对钴铈复合氧化物催化剂催化N2O分解的影响[J]. 物理化学学报,
2009,25,(6): 1033-1039.
doi: 10.3866/PKU.WHXB20090604LIU Chang, XUE Li, HE Hong. Influence of alkaline earth metals on cobalt-cerium composite oxide catalysts for N2O decomposition[J]. Acta Phys Chim Sin,
2009, 25(6):
1033-1039.
doi: 10.3866/PKU.WHXB20090604
-
[23]
ZHANG Q L, TANG X S, NING P, DUAN Y K, SONG Z X, SHI Y Z. Enhancement of N2O catalytic decomposition over Ca modified Co3O4 catalyst[J]. Rsc Adv,
2015, 5(63):
51263-51270.
doi: 10.1039/C5RA04062K
-
[24]
郑丽, 吴藏藏, 徐秀峰. N2O在Mg-Co和Mg-Mn-Co复合氧化物上的催化分解[J]. 燃料化学学报,
2016,44,(12): 1494-1501.
doi: 10.3969/j.issn.0253-2409.2016.12.013ZHEN Li, WU Cang-cang, XU Xiu-feng. Catalytic decomposition of N2O over Mg-Co and Mg-Mn-Co composite oxides[J]. J Fuel Chem Technol,
2016, 44(12):
1494-1501.
doi: 10.3969/j.issn.0253-2409.2016.12.013
-
[25]
YU H B, WANG X P. Apparent activation energies and reaction rates of N2O decomposition via different routes over Co3O4[J]. Catal Commun,
2017, 106:
40-43.
-
[26]
KIM M J, LEE S J, RYU I S, JEON M W, MOON S H, ROH H S, JEON S G. Catalytic decomposition of N2O over cobalt based spinel oxides:The role of additives[J]. Mol Catal,
2017, 442:
202-207.
doi: 10.1016/j.mcat.2017.05.029
-
[27]
IVANOVA Y A, SUTORMINA E F, ISUPOVA I A, VONK E I. Catalytic activity of the oxide catalysts based on Ni0.75Co2.25O4 modified with cesium cations in a reaction of N2O decomposition[J]. Kinet Catal,
2017, 58(6):
793-799.
-
[28]
QU Z P, GAO K, FU Q, QIN Y. Low-temperature catalytic oxidation of toluene over nanocrystal-like Mn-Co oxides prepared by two-step hydrothermal method[J]. Catal Commun,
2014, 52:
31-35.
doi: 10.1016/j.catcom.2014.03.035
-
[29]
BIN F, SONG C L, LÜ G, SONG J O, CAO X F, PANG H T, WANG K P. Structural characterization and selective catalytic reduction of nitrogen oxides with ammonia:A comparison between Co/ZSM-5 and Co/SBA-15[J]. J Phys Chem C,
2012, 116:
26262-26274.
doi: 10.1021/jp303830x
-
[30]
XIE P F, LUO Y J, MAQ Z, WANG L Y, HUANG C Y, YUE Y H, HUA W M, GAO Z. CoZSM-11 catalysts for N2O decomposition:Effect of preparation methods and nature of active sites[J]. Appl Catal B:Environ,
2015, 170:
34-42.
-
[31]
武海鹏, 冯鸣, 徐秀峰. K改性Ni-Co-Al三元复合氧化物催化分解N2O[J]. 燃料化学学报,
2012,40,(7): 872-877.
doi: 10.3969/j.issn.0253-2409.2012.07.017WU Hai-peng, FENG Ming, XU Xiu-feng. Catalytic decomposition of N2O over potassium promoted Ni-Co-Al ternary mixed oxides[J]. J Fuel Chem Technol,
2012, 40(7):
872-877.
doi: 10.3969/j.issn.0253-2409.2012.07.017
-
[32]
吴藏藏, 张海杰, 王建, 徐秀峰. N2O分解催化剂Co-Al尖晶石型复合氧化物制备参数的优化[J]. 分子催化,
2016,30,(1): 62-71.
WU Cang-cang, ZHANG Hai-jie, WANG Jian, XU Xiu-feng. The preparation parameters screening of Co-Al spinel oxides for N2O catalytic decomposition[J]. J Mol Catal (China),
2016, 30(1):
62-71.
-
[33]
LI X, YANG Z C, QI W, LI Y T, WU Y, ZHOUu S X, HUANG S, WEI J, LI H J, YAO P. Binder-free Co3O4@NiCoAl-layered double hydroxide core-shell hybrid architectural nanowire arrays with enhanced electrochemical performance[J]. Appl Surf Sci,
2016, 363:
381-388.
doi: 10.1016/j.apsusc.2015.12.039
-
[34]
LÜ L, SU Y G, LIU X Q, ZHENG H Y, WANG X J. Synthesis of cellular-like Co3O4 nanocrystals with controlled structural, electronic and catalytic properties[J]. J Alloy Compd,
2013, 553:
163-166.
doi: 10.1016/j.jallcom.2012.10.164
-
[35]
WU Z X, DENG J G, LIU Y X, XIE S H, JIANG Y, ZHAO X T, YANG J, ARANDIYA NRABDIYAN H, GUO G S, DAI H X. Three-dimensionally ordered mesoporous Co3O4-supported Au-Pd alloy nanoparticles:High-performance catalysts for methane combustion[J]. J Catal,
2015, 332:
13-24.
doi: 10.1016/j.jcat.2015.09.008
-
[36]
YAN Z X, XU Z H, CHENG B, JIANG C J. Co3O4 nanorod-supported Pt with enhanced performance for catalytic HCHO oxidation at room temperature[J]. Appl Surf Sci,
2017, 404:
426-434.
doi: 10.1016/j.apsusc.2017.02.010
-
[37]
WANG Z, WANG W Z, ZHANG L, JIANG D. Surface oxygen vacancies on Co3O4 mediated catalytic formaldehyde oxidation at room temperature[J]. Catal Sci Technol,
2016, 6(11):
3845-3853.
doi: 10.1039/C5CY01709B
-
[38]
YU, WANG, WU, CH, Y. Promotion of Ag for Co3O4 catalyzing N2O decomposition under simulated real reaction conditions[J]. Chem Eng J,
2018, 334:
800-806.
doi: 10.1016/j.cej.2017.10.079

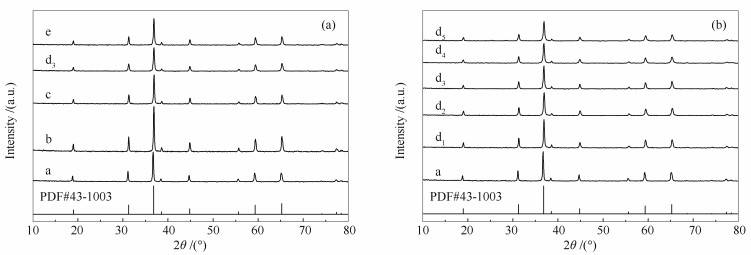
 下载:
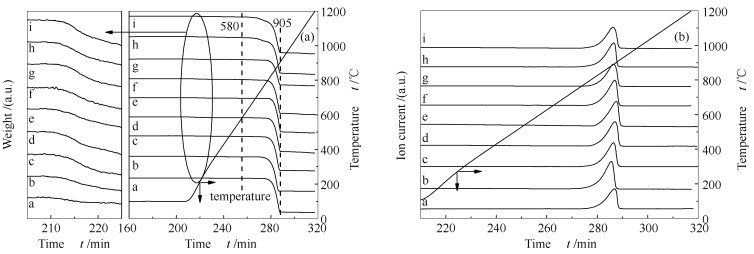
下载:

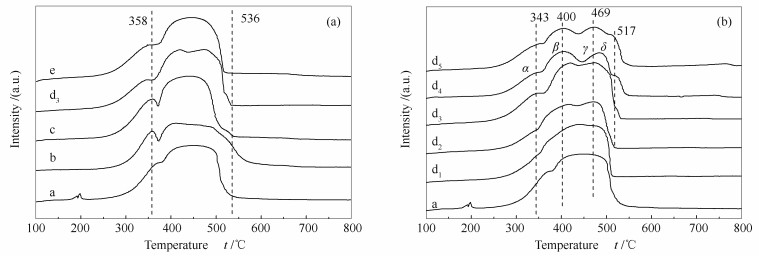

 下载:
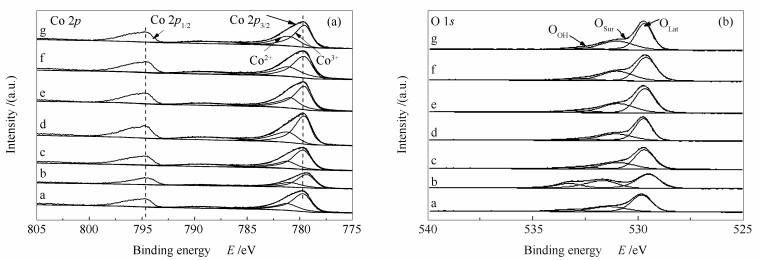
下载:







 下载:
下载:
