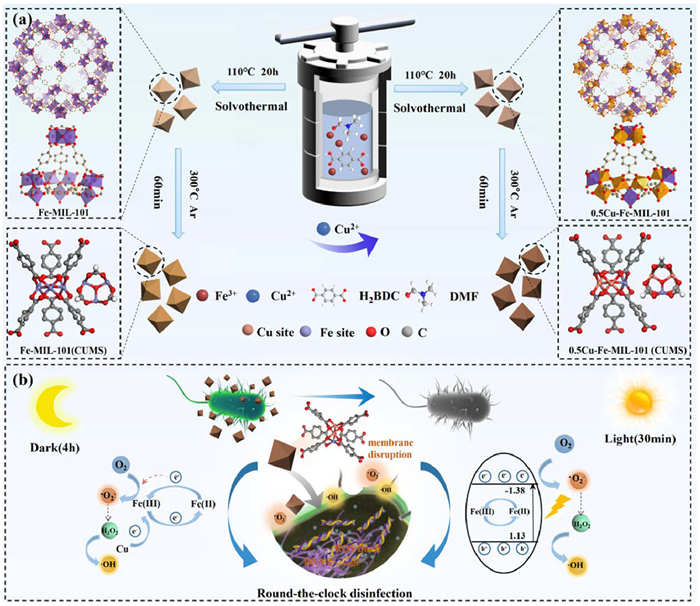
Scheme 1.
Schematic illustration of (a) the preparation process of Fe-MIL-101(CUMS) and Cu-doped Fe-MIL-101(CUMS) and (b) day-night antimicrobial mechanism.
Regulating electron transfer between valence-variable Fe and Cu sites in bimetallic MOFs to enhance ROS generation for day-night antibacterial efficacy
Shanshan Cheng , Lingjing Yu , Siqi Liu , Hongyi Gao , Changan Wang , Zeyang Song , Supakorn Boonyuen , Muye Niu , Ge Wang

Pathogenic infections caused by bacteria and viruses pose significant threats to human health and development [1]. Traditional bactericidal mechanisms employed by nano-antimicrobial materials, such as metal ion dissolution, surface physical puncture, and photothermal lysis, have been extensively studied [2,3]. Recently, reactive oxygen species (ROS) mediated oxidative stress has emerged as a novel and extensively researched antimicrobial mechanism. ROS possess strong oxidizing properties, which include hydroxyl radicals (·OH), superoxide radicals (·O2−), hydrogen peroxide (H2O2), and singlet oxygen (1O2). These species are primarily generated through the interaction of high-energy carriers on the surface of antimicrobial materials with O2, H2O, and other elements via electron transfer and related processes. As antimicrobial reaction intermediates, ROS can react with the cell membranes, proteins, and other components of pathogens, causing oxidative stress and pathogen death, which is a highly efficient active ingredient of antimicrobial agents [4,5]. Currently, many nanomaterials (TiO2 [6], ZnO [7], and black phosphorus [8]) can generate ROS with the assistance of exogenous stimuli, including light, ultrasound, and endogenous chemicals like H2O2. The materials that release ROS through photodynamic, thermodynamic, and photocatalytic interventions have been extensively used in the antimicrobial field [9,10]. However, the reliance on external stimuli for ROS generation significantly limits their practical utility. Therefore, it is essential to develop nanomaterials that can generate ROS through catalytic oxidation without external triggers.
Metal-organic frameworks (MOFs) are porous materials characterized by a regular topology, formed through the coordination of metal ions or clusters with organic ligands. Their low toxicity and excellent biodegradability have attracted significant attention for medicine and biology applications, particularly as novel antimicrobial agents [11,12]. The high specific surface area, tunable porous structure, and abundant nanochannels of MOFs facilitate the efficient transfer of guest molecules such as O2 and H2O2 during reactions [13]. Previous studies have demonstrated the exceptional photocatalytic bactericidal activity of zeolitic imidazolate framework-8 (ZIF-8), which generates photogenerated electrons via ligand-to-metal charge transfer (LMCT) upon light exposure. The generated ·O2− and H2O2 can effectively oxidize and eliminate pathogens [14]. The size of 2D PCN-134 MOF nanosheets influences their photodynamic properties, with increased photodynamic activity for ROS generation observed as the size decreases [15]. Furthermore, Fe-MOFs derivative catalysts prepared using MIL-100(Fe) as a precursor and CuS-modified MIL-101(Fe), exhibited impressive 1O2-mediated antibacterial activity against Escherichia coli under visible light [16,17]. However, the bactericidal effect diminishes significantly in the dark. It can be seen that MOFs catalyze ROS generation to achieve antimicrobial activity somewhat controlled by the light-driven reaction, the ability to activate O2 to produce ROS in the dark is relatively limited. Meanwhile, the MOFs depend on a light-driven mechanism that presents problems such as wide bandgap, poor visible light absorption, low carrier separation efficiency, and slow charge transfer due to poor electron conductivity [18,19]. This limits their antimicrobial capacity during diurnal processes. Addressing this challenge is crucial for improving the antimicrobial efficacy of MOFs throughout the diurnal cycle.
Recent studies have revealed that certain metal nanoparticles and metal oxides can enhance O2 adsorption and promote ROS generation in the dark through improved electron transfer. These strategies typically involve the modulation of surface structures, surface defects, particle size, and the synergistic effects of various components to facilitate electron transfer [20–24]. Achieving precise control of ROS generation and improvement of ROS production efficiency under dark conditions is possible by modulating the electron transfer of MOFs and designing abundant and homogeneous metal active sites. For instance, previous research demonstrated that Zn@MOFs can spontaneously release ·O2− and H2O2 via electron transfer from the zinc core to the MOF shell, resulting in significant antimicrobial efficacy against various pathogens [25]. In contrast, bimetallic sites modulation can promote the spontaneous generation of ROS by optimizing the metal active sites, modulating the electronic structure, and mimicking the activity of the nano-enzymes [26,27]. The charge transfer during spontaneous ROS generation was facilitated by modulation of the metal active sites of Cu-doped ZIF-8, which generates ·O2−, ·OH in the dark [28]. The bimetallic synergistic effect can reduce the bandgap, optimize the energy band structure, and broaden the photo-response range through intermetallic electron transfer or orbital hybridization [29]. Meanwhile, the design of coordinatively unsaturated metal sites (CUMS) in MOFs can expose highly active sites and facilitate charge separation. CUMS act as electron traps to capture photo-generated electrons and inhibit complexation. The electron conductivity can be enhanced by the low-coordination state of the metal centers, which can also maintain and drive the chemocatalytic cycle (e.g., Fenton reactions) to generate ROS in the dark, reducing dependence on the light [30,31]. The active sites of MIL-101(Fe) can be engineered to generate CUMS, thereby facilitating efficient ·O2− and ·OH mediated antimicrobial activity in the dark [32]. The concept of achieving round-the-clock antibacterial activity through the modulation of metal active sites and electron transfer is promising.
In this study, we leverage the precise and tunable structures and generation of valence-variable metal sites of Fe-MOFs, allowing for the rational modulation of the electronic microenvironment around Fe sites through Cu incorporation and thermal activation, leading to the formation of abundant CUMS. We systematically investigated the synergistic effects of electronic structure modulation on O2 activation and continuous ROS generation. The higher electronegativity of Cu significantly accelerates the regeneration of valence-variable Fe(Ⅱ) sites and optimizes the electronic structure of CUMS in the bimetallic MOFs, thereby enhancing the valence conversion processes [33]. The development of CUMS not only optimizes electron distribution among valence-variable Fe sites but also strengthens the interaction of valence-variable sites with H2O2, facilitating O2 adsorption and activation. The integration of CUMS and transition metal Cu sites in Fe-MOFs synergistically optimize the energy band structure, active sites and electron transfer, boosting the catalytic efficiency for ROS generation and sterilization. Consequently, the 0.5Cu-Fe-MIL-101(CUMS) demonstrated exceptional ROS generation capacity, achieving an antibacterial efficacy of 99.99% against Gram-negative E. coli under day-night conditions. density functional theory (DFT) calculations confirmed that enhanced electron transfer between Cu and Fe sites promotes the adsorption of O2 and H2O2 on the valence-variable Fe sites, resulting in the spontaneous generation of ·O2− and ·OH, which induce oxidative stress of bacterial cells. This study underscores the potential of transition metal doping and the introduction of CUMS in optimizing electron transfer in Fe-MOFs, providing valuable insights for the development of innovative day-night antibacterial catalysts.
As illustrated in Scheme 1, a series of Cu-doped Fe-MIL-101(CUMS) nanoparticles were prepared by Fe(Ⅲ) and Cu(Ⅱ) precursors at different molar ratios of 1:9, 1:3, 3:7, and 1:1. The synthesis involved the reaction of Fe(Ⅲ) ions with the -COOH of H2BDC at a temperature of 110 ℃, resulting in a notable color transition of the products from light orange to darker orange with the incremental introduction of Cu(Ⅱ) ions (Figs. S3a–e in Supporting information). Subsequently, the Cu-doped samples were thermally treated in an Ar-protected tube furnace at 300 ℃, yielding xCu-Fe-MIL-101(CUMS) with coordinatively unsaturated metal sites. Notably, the color shifted from orange to brown following the heat treatment, with the intensity of the brown color increasing as the level of Cu doping (Figs. S3f–j in Supporting information).
The scanning electron microscopy (SEM) image demonstrates that Fe-MIL-101 exhibits a smooth octahedral morphology, characterized by well-defined corners and a diameter ranging from approximately 500 nm to 1 μm (Fig. 1a), which is consistent with the literature [34]. Upon doping with Cu, a notable reduction in the particle size of xCu-Fe-MIL-101 was observed, correlating with increased Cu content (Fig. S4 in Supporting information). The morphology transitioned from a regular to a more irregular octahedral shape, with less distinct angles. This alteration is attributed to the substitution of Fe sites by Cu ions. The larger ionic radius of Cu(Ⅱ) compared to Fe(Ⅲ) facilitates greater spatial occupation within the crystal lattice, modifying the coordination environment. Furthermore, this substitution may influence the configuration and stability of the organic linkers, potentially impeding the growth of crystal grain [35,36]. Conversely, the Fe-MIL-101 and xCu-Fe-MIL-101(CUMS) (x = 0.1, 0.25, 0.3, 0.5) maintained their octahedral structure, exhibiting changes in particle size and the surface became roughened (Figs. 1b–f). TEM images further illustrated uniformly distributed voids within the samples while preserving the integrity of the octahedral framework (Figs. 1g–i). The uniform distribution of the Cu, Fe, C, and O elements in both Fe-MIL-101(CUMS) and xCu-Fe-MIL-101(CUMS) (x = 0.25, 0.5) was further confirmed by energy dispersive X-ray spectroscopy (EDX) (Fig. 1j and Fig. S5 in Supporting information).

The electronic structure, electron transfer at the coordinatively unsaturated bimetallic sites, and the influence of Cu doping on the electronic structure of xCu-Fe-MIL-101(CUMS) were investigated by X-ray photoelectron spectroscopy (XPS). The XPS analysis of 0.5Cu-Fe-MIL-101(CUMS) (Fig. 2a) confirmed the presence of C, O, Fe, and Cu, indicating the Cu content gradually increased and the Cl content decreased following Cu doping and subsequent heat treatment (Table S5 in Supporting information), which further substantiates the successful incorporation of Cu and the elimination of Cl− at 300 ℃. The Fe 2p XPS spectra of Fe-MIL-101 reveal peaks at 711.5 and 724.6 eV, corresponding to Fe 2p3/2 and Fe 2p1/2, respectively (Fig. 2b). The peaks at 711.5, 715.41, 724.6, and 727.74 eV are indicative of Fe(Ⅲ), while those at 719.21 and 730.75 eV represent Fe(Ⅲ) fingerprints [37,38]. The Fe 2p spectrum of Fe-MIL-101(CUMS) exhibited a notable shift towards lower binding energies, with peaks at 711.4 and 724.58 eV. Additionally, a new peak at 710.5 eV was attributed to the presence of Fe(Ⅱ), which was generated in CUMS following heat treatment. The Fe 2p spectra of 0.25Cu-Fe-MIL-101(CUMS) and 0.5Cu-Fe-MIL-101(CUMS) exhibited further shifts towards lower binding energies under consistent temperature and time of heat treatment, with values of 711.30 and 724.50 eV, and 711.19 and 724.45 eV, respectively. Furthermore, new peaks at 710.40 and 710.25 eV were attributed to Fe(Ⅱ) generated from Cu doping and the formation of CUMS after heat treatment.
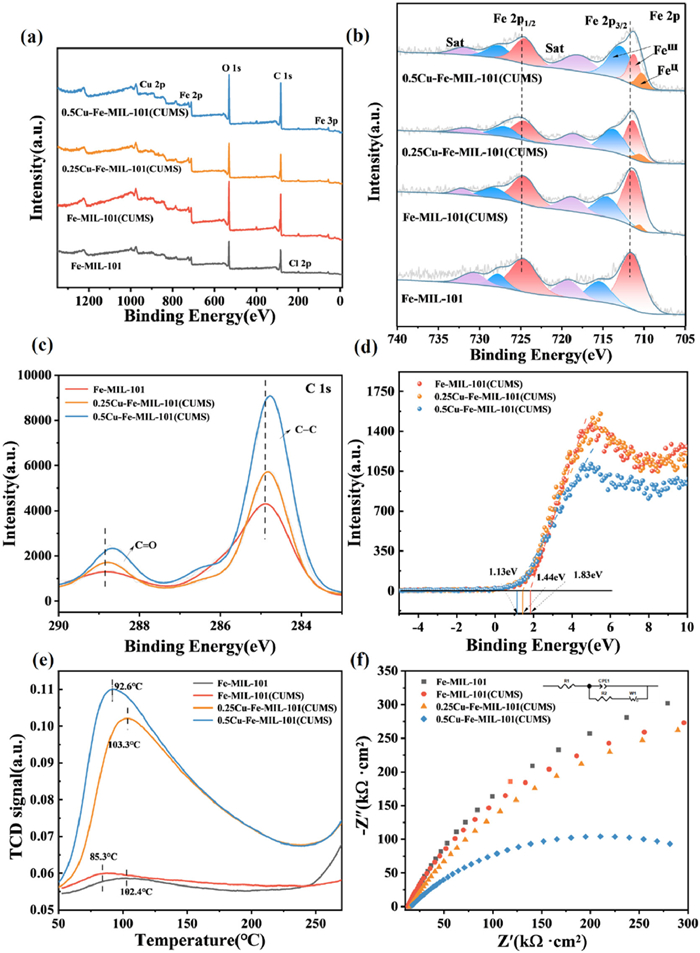
The binding energies of Fe 2p3/2 and Fe 2p1/2 for the last three samples exhibit a notable decrease relative to those in Fe-MIL-101, indicating a negative energy shift. This binding energy shift increases slightly with higher Cu doping. The 0.5Cu-Fe-MIL-101(CUMS) demonstrates the most pronounced shift, suggesting that the introduction of Cu sites enhances the electron density surrounding the Fe atoms. The interaction between the Fe and Cu sites in 0.5Cu-Fe-MIL-101(CUMS) facilitates electron transfer from Cu to Fe, thereby reducing the binding energy of Fe 2p This suggests that Cu acts as an electron donor to modulate the electronic structure of Fe sites to form highly reactive CUMS. The ratios of Fe(Ⅱ) to Fe(Ⅲ) significantly increase to 0%, 1.12%, 3.99%, and 6.19% for the respective samples (Table S5), indicating that heat treatment effectively removes H2O molecules and ligand anions, resulting in the formation of CUMS, while simultaneously allowing partial reduction of Fe(Ⅲ) to Fe(Ⅱ). The presence of Cu sites alters the chemical environment surrounding the Fe(Ⅲ) sites. Meanwhile, spontaneous charge transfer from Cu to Fe(Ⅲ) may occur, given that the conversion of Cu(Ⅱ) to Cu(Ⅰ) (E = 0.16 V vs. normal hydrogen electrode (NHE)) is more favorable than the reduction of Fe(Ⅲ) to Fe(Ⅱ) (E = 0.77 V vs. NHE) [39]. Thus, the Cu sites potentially enhance the generation of Fe(Ⅱ).
In comparison to Fe-MIL-101, the C 1s spectra of 0.25Cu-Fe-MIL-101(CUMS) and 0.5Cu-Fe-MIL-101(CUMS) exhibited a notable shift towards lower binding energies (Fig. 2c). This shift suggests that the coordination environment of the C atoms with the metal centers was altered. The electronic interaction between the metal centers and the organic ligands appears to facilitate the reduction of the C atoms, driven by the electronic coupling effect. These findings indicate that Cu doping enhances electron transfer between the metal sites and ligands through cation-π interactions.
The shift of the valence band spectrum towards higher binding energies of the catalysts is associated with alterations in the electronic structure [40]. The VB spectrum of 0.5Cu-Fe-MIL-101(CUMS) demonstrates a blue shift towards the vacuum level compared to Fe-MIL-101(CUMS) (Fig. 2d), confirming that Cu doping results in the modification of the electronic structure of MOFs [41]. The intersection with the horizontal line obtained by tangent fitting to the spectrogram corresponds to the theoretical valence band of 1.83, 1.44, and 1.13 eV, respectively, indicating a reduction in valence band energy. This reduction can be attributed to the substitution of Fe sites by Cu, which modifies the electronic arrangement of the metal centers in 0.5Cu-Fe-MIL-101(CUMS), thereby affecting the overall electronic structure of the material.
The relationship between Lewis acidic sites and CUMS in MOFs is significant. Typically, the metal centers or clusters within MOFs exhibit Lewis acidity, and the presence of CUMS substantially enhances Lewis acidity [42,43]. The CUMS serve as catalytically active centers, offering additional electron-acceptor sites and enhancing interaction capabilities with reactant molecules [44]. The samples displayed distinct desorption peaks (Fig. 2e) in the range of 50–250 ℃. The peaks around 100 ℃ correspond to weak acidic sites, while those between 300 ℃ and 500 ℃ indicate moderate acidic sites. The adsorption peak of 0.5Cu-Fe-MIL-101(CUMS) was located at 92.6 ℃, corresponding to Lewis acidic sites, with the low-temperature desorption peak indicating a weak acidic center. The area of the desorption peak reflects the number of acidic sites corresponding to acid strengths, The larger peak area signifies the greater quantity of acidic sites [45,46]. Following heat treatment, the adsorption peaks associated with weak acids were significantly enhanced. Notably, 0.5Cu-Fe-MIL-101(CUMS) exhibited the highest adsorption peak intensity and the largest peak area, indicating that it possesses the greatest number of Lewis acid sites and a higher concentration of CUMS.
The electron transfer of the samples were evaluated by electrochemical impedance spectra (EIS) [47]. Notably, the arc radius in the EIS Nyquist plots progressively decreases as Cu doping increases compared to that of Fe-MIL-101, with the smallest arc radius observed in 0.5Cu-Fe-MIL-101(CUMS) (Fig. 2f). EIS analysis suggests that the incorporation of Cu sites in 0.5Cu-Fe-MIL-101(CUMS) accelerates the interfacial charge transfer rate, while concurrently reducing charge transfer resistance. These findings underscore the role of Cu sites in reducing electron transfer resistance and significantly enhancing interfacial electron transfer.
To investigate the in vitro antimicrobial activity of the materials, Fe-MIL-101 and xCu-Fe-MIL-101(CUMS) (x = 0, 0.1, 0.25, 0.3, 0.5) were evaluated for their growth inhibition of E. coli in the dark (Fig. S8 in Supporting information). The relative viabilities of E. coli treated with different concentrations of samples under dark conditions were determined (Fig. 3a). The bacterial relative viability of 0.25Cu-Fe-MIL-101(CUMS) and 0.5Cu-Fe-MIL-101(CUMS) was significantly lower compared to the samples without Cu doping, both of which killed all bacteria at concentrations of 250 and 100 μg/mL, respectively. The antimicrobial activity of Fe-MIL-101 and Fe-MIL-101(CUMS) was limited at a concentration of 100 μg/mL, killing only 54.39% and 59.27% of E. coli, respectively. In contrast, the xCu-Fe-MIL-101(CUMS) (x = 0.1, 0.25, 0.3, 0.5) showed antimicrobial activity against E. coli of 93.63%, 98.31%, 99.57%, and 99.99%, respectively (Fig. 3b). Notably, 0.5Cu-Fe-MIL-101(CUMS) demonstrated superior antibacterial properties against E. coli, indicating that the incorporation of Cu sites significantly enhanced the overall antibacterial activity. To accurately verify the antimicrobial effect of 0.5Cu-Fe-MIL-101(CUMS) at low concentrations, the concentration range was adjusted to 0.291–12.50 μg/mL. The half-maximal inhibitory concentration (IC50) of 0.5Cu-Fe-MIL-101(CUMS) was tested to be 1.453 μg/mL (Fig. S9 in Supporting information), which provides a more accurate indicator of activity for future applications.
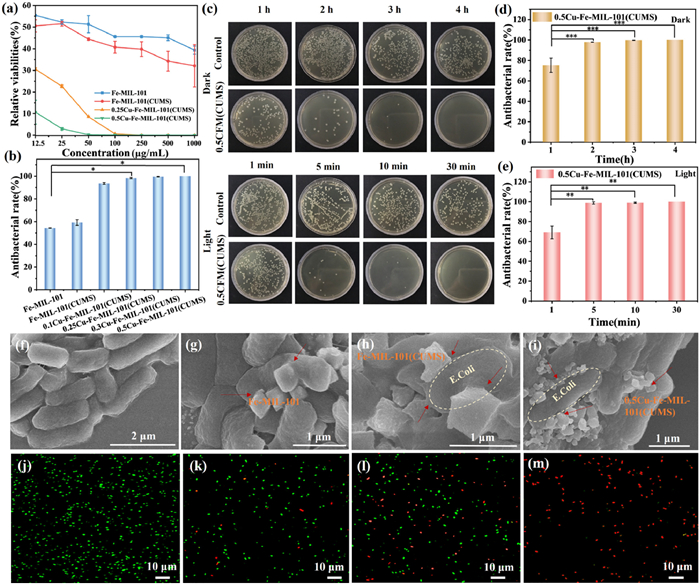
Plate photographs illustrated a significant decrease in colony counts over time, with the most pronounced antibacterial effect at 4 h in the dark (Fig. 3c). The antibacterial rate of 0.5Cu-Fe-MIL-101(CUMS) (100 μg/mL) against E. coli was 75.37%, 97.94%, 99.65%, and 99.99% after 1, 2, 3, and 4 h treatment under darkness (Fig. 3d), respectively. The MBC of 0.5Cu-Fe-MIL-101(CUMS) was determined to be 100 μg/mL in the dark (Fig. S10 in Supporting information). Furthermore, the antimicrobial performance of 0.5Cu-Fe-MIL-101(CUMS) was evaluated under light conditions, yielding antibacterial rates of 69.02%, 98.78%, 98.94%, and 99.99% after 1, 5, 10, and 30 min, respectively (Fig. 3e). Colony counts showed a significant reduction after 5 min of exposure to light, with complete bacterial death after 30 min (Fig. 3c). The MBC was determined to be 250 µg/mL after 30 min under light conditions (Figs. S10 and S11 in Supporting information). Notably, the 0.5Cu-Fe-MIL-101(CUMS) effectively killed E. coli in the dark and light. Compared to the antimicrobial properties of antimicrobial catalysts mentioned in previous studies, the 0.5Cu-Fe-MIL-101(CUMS) catalysts synthesized in this work demonstrated competitive performance under all-weather conditions. The antibacterial performance of catalysts with those reported compared and summarized in Table S6 (Supporting information).
The membrane damage and cell wall disruption of the NPs-treated bacteria were observed by SEM. It was evident that the untreated control E. coli was well-preserved rod shape with a normal appearance, and the cell wall remained intact (Fig. 3f). After the addition of Fe-MIL-101 NPs, the materials mostly adhered to the cell surface of E. coli and were accompanied by octahedral NPs puncturing the cell wall (Fig. 3g). There was deformation and wrinkling of the bacterial cell wall, indicating that the Fe-MIL-101 NPs had a certain antimicrobial effect on the E. coli. The state of E. coli after interaction with Fe-MIL-101(CUMS) is similar to the effect of Fe-MIL-101 (Fig. 3h). When exposed to 0.5Cu-Fe-MIL-101(CUMS) NPs, the cell walls of E. coli displayed significant depressions and the bacterial cell membranes were disrupted (Fig. 3i). The crumpled and contracted appearance of the cell walls further suggests that 0.5Cu-Fe-MIL-101(CUMS) possesses enhanced antibacterial activity.
The interaction of MOFs with bacteria was investigated by CLSM to visually assess the disruption of bacterial cell walls. Bacterial cells were stained with SYTO 9 and propidium iodide (PI), E. coli of the control group displayed strong green fluorescence, which exhibited intact cell walls (Fig. 3j). The slightly red fluorescence was detected following treatment with Fe-MIL-101, suggesting minor cell damage (Fig. 3k). In contrast, the green fluorescence intensity decreased significantly after treatment with Fe-MIL-101(CUMS), indicating the increase of bacterial cell death (Fig. 3l). Remarkably, E. coli treated with 0.5Cu-Fe-MIL-101(CUMS) exhibited predominantly red fluorescence (Fig. 3m), signifying substantial damage to the bacterial cell wall and a high rate of cell death due to exposure to catalysts. These findings corroborate the results of in vitro antimicrobial experiments, demonstrating that the incorporation of Cu sites into Fe-MIL-101 significantly enhances antimicrobial activity.
Previous studies indicated that the E. coli cell surfaces exhibit a negative charge within a pH range of 3–11 [48,49]. The zeta potential of Fe-MIL-101 suspended in phosphate buffer saline (PBS) at pH 7 was measured as 5.91 mV. In contrast, the zeta potentials of Fe-MIL-101(CUMS) and 0.5Cu-Fe-MIL-101(CUMS) were tested to be −9.21 and −2.79 mV, respectively (Fig. 4a). The negative zeta potential of xCu-Fe-MIL-101(CUMS) compared to Fe-MIL-101 suggests an increased electron density at the surface of the catalysts. However, the electrostatic interaction diminishes due to the electrostatic repulsion between the nanoparticles and bacteria cells, which excludes the contribution of electrostatic interactions to the antibacterial effect.
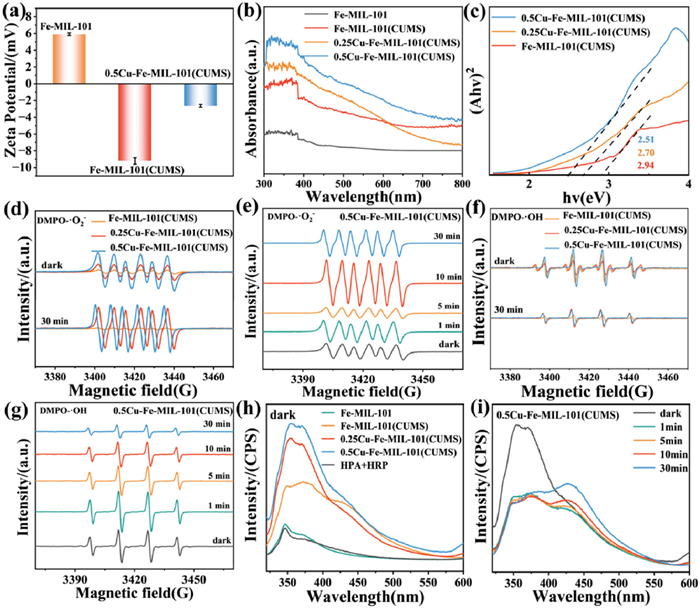
To explore the sterilization capability of the materials under light conditions, we analyzed the optical properties using ultraviolet-visible (UV–vis) spectroscopy (Fig. 4b), which revealed that incorporating Cu sites and subsequently thermally treating the materials significantly increases the photo-responsive wavelength range (380–750 nm). Both modified materials demonstrated significant absorption in the visible region, suggesting their potential for visible light utilization. With Cu doping increased, the intensity of visible light absorption of the catalysts progressively enhanced. The 0.5Cu-Fe-MIL-101(CUMS) exhibits the most pronounced ability to capture visible light, which is beneficial to the photocatalytic performance. The abundant Fe-O clusters in Fe-MIL-101 effectively broaden the light absorption spectrum of the visible range [50]. Additionally, the thermal treatment promotes the exposure of unsaturated Fe(Ⅱ) active sites, which in conjunction with the Cu sites, significantly enhances the photogenerated electron transfer and facilitates the oxygen reduction process in the light. This synergy accelerates the reduction of Fe(Ⅲ) to Fe(Ⅱ), improving the overall efficiency of the photocatalytic oxidation reaction.
The band gap energies of samples were analyzed (Fig. 4c). The band gap energy of Fe-MIL-101 was 3.06 eV (Fig. S12 in Supporting information), while the band gap energy of 0.5Cu-Fe-MIL-101(CUMS) decreases to 2.51 eV with the Cu doping and thermal treatment, which contributes to the enhanced charge separation and transfer [51]. It has been demonstrated that introducing Cu sites effectively increases the light absorption capacity of Fe-MOFs in the visible region.
The corresponding ROS (·O2−, ·OH and 1O2) generation of samples in the dark and light was detected using 5, 5-dimethyl-1-pyrroline N-oxide (DMPO)/2, 2, 6, 6-tetramethyl-4-piperidone hydrochloride (TEMP) as a spin agent by EPR. For the mixture of DMPO and the different samples, the absorption signals of typical DMPO-·O2− adducts were tested using methanol as solvent (Fig. 4d). DMPO-·O2− adducts with six lines hyperfine pattern (hyperfine splitting constants: aN = 13.6 G and aH = 7.7 G) was identified as the dominant adducts, indicating that the materials produce accumulated ·O2− [52]. The intensity of the adduct signal increases gradually with the increase of Cu content, indicating that the 0.5Cu-Fe-MIL-101(CUMS) is capable of producing more ·O2− under dark and light conditions, with the highest efficiency to generate ·O2−. The stable generation of ·O2− was observed at 30 min of light illumination (Fig. S13a in Supporting information). The 0.5Cu-Fe-MIL-101(CUMS) was investigated under different conditions, which is demonstrated to be the most effective in generating ·O2− (Fig. 4e). It was observed that the ·O2− produced by 30 min of light was greater than that produced in the dark. The ·O2− produced by the materials was determined by detecting the consumption of nitroblue tetrazolium (NBT). The samples produce ·O2− in the dark and light, with 0.5Cu-Fe-MIL-101(CUMS) demonstrating a greater capacity to produce ·O2− than Fe-MIL-101 and Fe-MIL-101(CUMS) (Figs. S13b and c in Supporting information), in accordance with the EPR results. The UV–vis spectra of 0.5Cu-Fe-MIL-101(CUMS) further indicate that light exposure of 30 min produced more ·O2− than that produced under dark conditions (Fig. S13d in Supporting information).
For the mixture of DMPO and the materials, typical signals of DMPO-·OH adducts (hyperfine splitting constants: aN = 15.1 G, aH = 14.8 G) were tested with deionized water as the solvent. The four characteristic peaks with intensity ratios of 1:2:2:1 could be identified as DMPO-·OH adducts, indicating that the catalysts produced accumulated ·OH [53]. The 0.5Cu-Fe-MIL-101(CUMS) exhibited the strongest adduct signal in the dark and light (Fig. 4f), and the intensity of the signal increased with the increase of Cu content. Compared with the other materials, 0.5Cu-Fe-MIL-101(CUMS) exhibits the greatest capacity for producing ·OH under dark and light conditions, demonstrating the highest efficiency in generating ·OH. In contrast to ·O2−, 0.5Cu-Fe-MIL-101(CUMS) produces more ·OH under dark conditions than under light for 30 min. Furthermore, the peak intensity decreases with increasing light time, resulting in a reduction in ·OH production (Fig. 4g and Fig. S14 in Supporting information). The ·OH can be captured by terephthalic acid (TPA) and reacts to form hydroxyterephthalic acid (TAOH), which is capable of emitting fluorescence. The samples produced ·OH in the dark and light, and the 0.5Cu-Fe-MIL-101(CUMS) can produce more ·OH than Fe-MIL-101(CUMS) and 0.25Cu-Fe-MIL-101(CUMS) (Figs. S14a and b in Supporting information).
The production of H2O2 of the samples under dark conditions was evaluated by fluorescence spectrophotometer with 4-Hydroxyphenylacetic acid (HPA) as the fluorescent probe (Fig. 4h). It can be observed that 0.5Cu-Fe-MIL-101(CUMS) is capable of producing more H2O2 than Fe-MIL-101 and Fe-MIL-101(CUMS), and the H2O2 produced by 0.5Cu-Fe-MIL-101(CUMS) under dark conditions is significantly higher than that produced by light for 30 min (Fig. 4i). This is consistent with the ·OH production by 0.5Cu-Fe-MIL-101(CUMS) in the dark as mentioned previously. Fe-MIL-101 contains numerous unsaturated Lewis acid Fe sites, which are capable of adsorbing the Lewis base H2O2 to produce ·OH via the Fenton reaction. The introduction of Cu sites in 0.5Cu-Fe-MIL-101(CUMS) may facilitate spontaneous charge transfer from the Cu sites to the Fe(Ⅲ) sites, accelerating the regeneration of Fe(Ⅱ) and improving the dissociation of H2O2 to produce ·OH.
The capacity of the catalysts to generate 1O2 was evaluated using TEMP as a trapping agent. Notably, there is an absence of 1O2 of the materials (Figs. S15a–c in Supporting information). It can be confirmed that the 0.5Cu-Fe-MIL-101(CUMS) exhibits the most effective all-weather generation of ·O2−, ·OH, and H2O2, which play a key role in antibacterial activity. Meanwhile, a positive correlation was observed between the Cu doping amount and enhanced photo-response, reduced bandgap, increased all-weather ROS generation and higher antimicrobial rates. This suggests that a suitable proportion of Cu doping enables Cu sites to fully enter the lattice, thereby influencing the electronic structure of coordinatively unsaturated valence-variable sites. Furthermore, increasing Cu doping narrows the band gap and introduces intermediate states, which can significantly enhance the energy band modulation and photo-responsive performance of the catalysts, which is conducive to photogenerated electron transfer and redox generation of ROS.
The visual representation of the key parameters influencing the bactericidal properties of the catalysts is illustrated in Fig. S16 (Supporting information), which provides a comparative analysis of the specific surface area, Fe(%), Cu(%) and Fe(Ⅱ)/Fe(Ⅲ) ratios among the xCu-Fe-MIL-101(CUMS). Notably, the 0.5Cu-Fe-MIL-101(CUMS) exhibited the highest values for several critical parameters, which demonstrated the most effective antimicrobial performance. Specifically, the percentages of the Fe(Ⅱ)/Fe(Ⅲ) ratio were positively correlated with enhanced antimicrobial activity. The highest Fe(Ⅱ)/Fe(Ⅲ) ratio was observed for the 0.5Cu-Fe-MIL-101(CUMS) (Table S5). On the one hand, adjusting the Fe(Ⅱ)/Fe(Ⅲ) ratio can change the energy band structure of MOFs and optimize visible light absorption. On the other hand, the mixed valence states of Fe(Ⅱ)/Fe(Ⅲ) form redox pairs in Fe-MOFs, which significantly affect the electron transfer efficiency and enhance the catalytic activity of the Fenton reaction. Fe(Ⅱ) catalyzes the generation of H2O2 to ·OH via the Fenton reaction, whereas Fe(Ⅲ) produces ·O2− via the Fenton-like reaction. The mixed-valence system of 0.5Cu-Fe-MIL-101(CUMS) (Fe(Ⅱ)/Fe(Ⅲ)=6.19%) enabled the formation of dual reaction centers, which synergistically boosted the yield of ROS (·OH, ·O2−), which corresponds to the ·OH and ·O2− producing effect of 0.5Cu-Fe-MIL-101(CUMS). The Fe(Ⅱ)/Fe(Ⅲ) ratio significantly enhanced the antimicrobial activity by synergistically modulating the electron transfer, ROS generation and catalytic activity of the samples.
Meanwhile, the uniform distribution of Cu and Fe elements in 0.5Cu-Fe-MIL-101(CUMS) directly enhances the antimicrobial performance by optimizing the stability of the structure, enhancing the synergistic effect of the active sites and promoting the generation of ROS. In contrast, the specific surface area does not significantly influence O2 activation and ROS generation. The radar chart effectively visualizes these relationships, highlighting the crucial roles of Cu doping and valence-variable metal sites in enhancing the antimicrobial potential of 0.5Cu-Fe-MIL-101(CUMS).
The synergistic antimicrobial mechanism was proposed for 0.5Cu-Fe-MIL-101(CUMS) under day-night conditions (Scheme 1). It can be postulated that 0.5Cu-Fe-MIL-101(CUMS) initially interacted with the bacterial outer membrane physically and then disrupted the cell membranes by generating exogenous ROS in the dark. The formation of coordinatively unsaturated Fe(Ⅱ) sites and electron transfer are facilitated by electron donor-acceptor synergism. The d-orbital high electron density of Cu(Ⅱ) (d10 configuration) can act as the electron donor to modulate the electronic structure of Fe(Ⅲ) through outer electron transfer, the d-orbital electrons of Fe(Ⅲ) are redistributed to form a more activated coordinated unsaturated Fe(Ⅱ) sites. This suggests that the charge transfer from Cu sites to Fe(Ⅲ) sites is spontaneous, which indirectly facilitates the reduction process from Fe(Ⅲ) to Fe(Ⅱ) and accelerates the regeneration of Fe(Ⅱ). These coordinated unsaturated Fe(Ⅱ) sites expose highly reactive metal centers that adsorb O2 by coordination interaction and physisorption. The empty d-orbital of Fe(Ⅱ) attracts the lone electrons pair of O2 to form σ-type coordination bonds. While Cu doping enhances the electron transfer ability of Fe(Ⅱ) sites, the electrons to activate O2 are derived from the loss of electrons from the Cu sites and the electrons lost from the oxidation reaction of Fe(Ⅱ) converted to Fe(Ⅲ). Electron transfer promotes O—O bond activation, making adsorbed O2 more receptive to electrons to form ·O2−. Meanwhile, the coordinated unsaturated Fe(Ⅱ) sites exhibit Lewis acidity and act as catalytically active centers capable of adsorbing the Lewis base H2O2. Fe(Ⅱ) transfers electrons to H2O2 to produce ·OH via the Fenton reaction. The Cu sites on 0.5Cu-Fe-MIL-101(CUMS) acted as an electron donor to regulate the electronic structure of Fe(Ⅲ), accelerating the regeneration of Fe(Ⅱ) and achieving efficient adsorption and activation of O2 and H2O2. The enhanced charge transfer at the Fe sites facilitates the adsorption of O2 and the generation of ROS, which disrupts the cell membrane and causes oxidative stress to the bacterial cells.
The energy band structure of Fe-MIL-101 is optimized by Cu doping and heat treatment, and the band gap energy of 0.5Cu-Fe-MIL-101(CUMS) is significantly reduced. The valence band electrons of 0.5Cu-Fe-MIL-101(CUMS) jump to the conduction band after photoexcitation, producing electron-hole pairs. These photogenerated carriers have high energies and can directly react with O2 adsorbed on Fe(Ⅱ) to reduce and generate ·O2−, which generates H2O2 through the following reaction. Meanwhile, the Fe(Ⅱ) sites undergo the Fenton reaction to generate ·OH. The generated ROS (·O2−, ·OH and H2O2) destroy the cell membrane, causing damage and oxidative stress to the bacterial cells and thus realizing efficient sterilization. The process of ROS generation is as follows.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
The above analysis proves that 0.5Cu-Fe-MIL-101(CUMS) exhibits antimicrobial properties through chemocatalysis-photocatalytic generation of ROS. In the absence of light, the redox cycle of the valence-variable Fe sites generates ROS through the chemocatalytic effect, which dominates as the basic mechanism. The chemically catalyzed ROS production plays a fundamental role and synergizes with the photocatalytic effect to significantly enhance the overall antimicrobial effect in the light, with photocatalytic generated ROS superimposed with chemocatalysis ROS to form a synergistic effect of oxidative stress.
In conclusion, a series of Cu-doped bimetallic xCu-Fe-MIL-101(CUMS) were successfully synthesized via a solvothermal method. The introduction of bimetallic sites and coordinatively unsaturated Fe sites was achieved by adjusting the Cu doping level and employing post-synthesis heat treatment. Notably, the ratio of Fe(Ⅱ)/Fe(Ⅲ) was influenced by Cu doping during the same heat treatment duration. Antibacterial assays demonstrated that 0.5Cu-Fe-MIL-101(CUMS) at a concentration of 100 μg/mL exhibited 99.99% antibacterial activity against E. coli under darkness for 4 h and light for 30 min, significantly surpassing that of Fe-MIL-101(CUMS). This material demonstrated the best day-night ROS production ability and bacterial inactivation effect. Experiments and DFT calculations revealed that Cu doping affects the electronic structure of the Fe sites and optimizes the energy band structure of Fe-MIL-101. The enhanced charge transfer on the Fe sites of 0.5Cu-Fe-MIL-101(CUMS) facilitates the adsorption of O2 and H2O2 for the spontaneous generation of ROS, which in turn promotes the production of ·O2− and ·OH. This process is critical for inducing oxidative stress and membrane damage in bacterial cells. This work provides valuable insights into the ROS-mediated oxidative stress mechanisms of Fe-MOFs, paving the way for their application in continuous antibacterial strategies.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Shanshan Cheng: Writing – review & editing, Writing – original draft, Visualization, Validation, Data curation. Lingjing Yu: Visualization, Software, Conceptualization. Siqi Liu: Validation. Hongyi Gao: Writing – review & editing, Supervision, Project administration, Funding acquisition. Changan Wang: Methodology. Zeyang Song: Formal analysis. Supakorn Boonyuen: Methodology. Muye Niu: Investigation. Ge Wang: Supervision, Resources, Funding acquisition.
This work was supported by the Beijing Natural Science Foundation (No. L233011) and Key Research and Development Projects of Hebei Province (No. 23313601D).
Supplementary material associated with this article can be found, in the online version, at doi:
L. Rizzello, P.P. Pompa, Chem. Soc. Rev. 43 (2014) 1501–1518. doi: 10.1039/C3CS60218D
Y.N. Slavin, J. Asnis, U.O. Häfeli, H. Bach, J. Nanobiotechnol. 15 (2017) 65. doi: 10.1186/s12951-017-0308-z
S. Duan, R.N. Wu, Y.H. Xiong, et al., Prog. Mater. Sci. 125 (2022) 100887. doi: 10.1016/j.pmatsci.2021.100887
H. Sies, V.V. Belousov, N.S. Chandel, et al., Nat. Rev. Mol. Cell Biol. 23 (2022) 499–515. doi: 10.1038/s41580-022-00456-z
M. Castro- Alférez, M.I. Polo-López, J. Marugán, P. Fernández-Ibáñez, Chem. Eng. J. 318 (2017) 214–223. doi: 10.1016/j.cej.2016.06.093
A. Kubacka, M.S. Diez, D. Rojo, et al., Sci. Rep. 4 (2014) 4134. doi: 10.1038/srep04134
H. Agarwal, S. Menon, S.V. Kumar, S. Rajeshkumar, Chem. Biol. Interact. 286 (2018) 60–70. doi: 10.1109/gucon.2018.8674988
L. Tan, J. Li, X.M. Liu, et al., Small 14 (2018) 1703197. doi: 10.1002/smll.201703197
W. Su, X.Y. Luo, P.Y. Li, et al., Chin. Chem. Lett. 35 (2024) 109522. doi: 10.1016/j.cclet.2024.109522
X.Y. Jin, F. Gao, M.X. Qin, et al., ACS Nano 16 (2022) 7755–7771. doi: 10.1021/acsnano.1c11647
A.C. McKinlay, R.E. Morris, P. Horcajada, et al., Angew. Chem. Int. Ed. 49 (2010) 6260–6266. doi: 10.1002/anie.201000048
Y.L. Li, X.Y. Zhang, S. Liu, et al., Chin. Chem. Lett. 36 (2025) 110501. doi: 10.1016/j.cclet.2024.110501
L. Qin, Z.W. Li, Z.H. Xu, et al., Appl. Catal. B: Environ. 179 (2015) 500–508. doi: 10.1016/j.apcatb.2015.06.001
P. Li, J.Z. Li, X. Feng, et al., Nat. Commun. 10 (2019) 2177. doi: 10.1038/s41467-019-10218-9
B.L. Xue, X.W. Geng, H.H. Cui, et al., Chin. Chem. Lett. 34 (2023) 108140. doi: 10.1016/j.cclet.2023.108140
J.X. Qin, Y. Pei, Y. Zheng, D.Q. Ye, Y. Hu, Appl. Catal. B: Environ. Energy. 325 (2023) 122346. doi: 10.1016/j.apcatb.2022.122346
Y.N. Jiang, Z.J. Wang, J.B. Huang, et al., Chem. Eng. J. 439 (2022) 135788. doi: 10.1016/j.cej.2022.135788
H. Luo, L. Yu, C. Liu, et al., J. Energy Chem. 103 (2025) 408–439. doi: 10.1016/j.jechem.2024.11.057
M.L. Ding, R.W. Flaig, H.L. Jiang, et al., Chem. Soc. Rev. 48 (2019) 2783–2828. doi: 10.1039/c8cs00829a
Q. Gao, Z.Y. Wang, Y.F. Rao, et al., J. Hazard. Mater. 443 (2023) 130275. doi: 10.1016/j.jhazmat.2022.130275
Z. Li, E.G. Wang, Y.Z. Zhang, et al., Nano Today 50 (2023) 101826. doi: 10.1016/j.nantod.2023.101826
V.L. Prasanna, R. Vijayaraghavan, Langmuir 31 (2015) 9155–9162. doi: 10.1021/acs.langmuir.5b02266
Y. Zhou, Y.F. Guo, J.Y. Li, et al., J. Mater. Chem. A 8 (2020) 11511–11514. doi: 10.1039/c9ta14044a
G.S. Yi, X.K. Li, Y. Yuan, Y.G. Zhang, Environ. Sci.: Nano 6 (2019) 68–74. doi: 10.1039/c8en01095a
J.Q. Wang, S.P. Teong, S.N. Riduan, et al., J. Am. Chem. Soc. 146 (2023) 599–608. doi: 10.1109/tr.2022.3192020
L.Y. Chen, H.F. Wang, C.X. Li, et al., Chem. Sci. 11 (2020) 5369–5403. doi: 10.1039/d0sc01432j
J. Bedia, V. Muelas-Ramos, M. Peñas-Garzón, et al., Catalysts 9 (2019) 52. doi: 10.3390/catal9010052
X.Z. Wang, H. Wang, J.F. Cheng, et al., Chem. Eng. J. 466 (2023) 143201. doi: 10.1016/j.cej.2023.143201
L.Z. Liu, J.C. Hu, Y. Sheng, et al., ACS Nano 18 (2024) 26271–26280.
Ü. Kökçam-Demir, A. Goldman, L. Esrafili, et al., Chem. Soc. Rev. 49 (2020) 2751–2798. doi: 10.1039/c9cs00609e
H.X. Li, Z.X. Yang, S. Lu, et al., Chemosphere 273 (2021) 129643. doi: 10.1016/j.chemosphere.2021.129643
S.Q. Peng, R. Li, Y.F. Rao, et al., Appl. Catal. B: Environ. 316 (2022) 121693. doi: 10.1016/j.apcatb.2022.121693
H. Liang, R.P. Liu, X.Q. An, et al., Chem. Eng. J. 414 (2021) 128669. doi: 10.1016/j.cej.2021.128669
A.D.S. Barbosa, D. Juliao, D.M. Fernandes, et al., Polyhedron 127 (2017) 464–470. doi: 10.1016/j.poly.2016.10.032
J.L. Han, X.D. He, J. Liu, et al., Chem 8 (2022) 1637–1657. doi: 10.1016/j.chempr.2022.03.006
Z.L. Huang, L.L. Fan, B. Chen, et al., J. Mater. Chem. A 9 (2021) 3976–3984. doi: 10.1039/d0ta08285f
T.Y. Yang, D.Y. Yu, D. Wang, et al., Appl. Catal. B: Environ. 286 (2021) 119859. doi: 10.1016/j.apcatb.2020.119859
J.T. Tang, J.L. Wang, Environ. Sci. Technol. 52 (2018) 5367–5377. doi: 10.1021/acs.est.8b00092
J.T. Tang, J.L. Wang, Chem. Eng. J. 375 (2019) 122007. doi: 10.1016/j.cej.2019.122007
F.M. Deng, Acta Phys. Sin. 65 (2016) 107101. doi: 10.7498/aps.65.107101
Z.Q. Xue, K. Liu, Q.L. Liu, et al., Nat. Commun. 10 (2019) 5048. doi: 10.1038/s41467-019-13051-2
R.F. Du, J.Y. Wu, Y.H. Fu, J. Adv. Phys. Chem. 7 (2018) 64–69. doi: 10.12677/JAPC.2018.72008
Z. Hu, D. Zhao, CrystEngComm 19 (2017) 4066–4081. doi: 10.1039/C6CE02660E
X.J. Wu, L. Li, L. Peng, et al., Acta Phys. Chim. Sin. 34 (2018) 286–295.
J.R. Grzechowiak, J. Rynowski, I. Wereszczako-Zieliñska, Catal. Today 65 (2001) 225–231. doi: 10.1016/S0920-5861(00)00562-9
L.L. Liu, X.J. Zhou, L. Liu, et al., Catalysts 9 (2019) 538. doi: 10.3390/catal9060538
F.X. Wang, Z.W. Zhang, F. Wang, et al., J. Colloid Interface Sci. 649 (2023) 384–393. doi: 10.1016/j.jcis.2023.06.083
S.O. Khelissa, M. Abdallah, C. Jama, et al., J. Mater. Environ. Sci. 8 (2017) 3326–3346.
C. Catania, A.W. Thomas, G.C. Bazan, Chem. Sci. 7 (2016) 2023–2029. doi: 10.1039/C5SC03046C
M.Y. Lan, X.W. Zhang, H.Y. Chu, C.C. Wang, Prog. Chem. 35 (2023) 458–474.
F.S. Farahani, M.S. Rahmanifar, A. Noori, et al., J. Am. Chem. Soc. 144 (2022) 3411–3428. doi: 10.1021/jacs.1c10963
J.F. Yan, J.L. Peng, L.D. Lai, et al., Environ. Sci. Technol. 52 (2018) 14302–14310. doi: 10.1021/acs.est.8b03340
S.Z. Pei, S.J. You, J. Ma, et al., Environ. Sci. Technol. 54 (2020) 13333–13343. doi: 10.1021/acs.est.0c05287

Scheme 1 Schematic illustration of (a) the preparation process of Fe-MIL-101(CUMS) and Cu-doped Fe-MIL-101(CUMS) and (b) day-night antimicrobial mechanism.

Figure 1 SEM images of (a) Fe-MIL-101, (b) Fe-MIL-101(CUMS) and (c, f) xCu-Fe-MIL-101(CUMS) (x = 0.1, 0.25, 0.3, 0.5). Low-magnification TEM image of (g) Fe-MIL-101(CUMS), (h) 0.25Cu-Fe-MIL-101(CUMS) and (i) 0.5Cu-Fe-MIL-101(CUMS). (j) The EDX elemental mappings images of 0.5Cu-Fe-MIL-101(CUMS).

Figure 2 (a) XPS survey spectra of Fe-MIL-101, xCu-Fe-MIL-101(CUMS) (x = 0, 0.25, 0.5). (b) High-resolution XPS spectra of Fe 2p of Fe-MIL-101, xCu-Fe-MIL-101(CUMS) (x = 0, 0.25, 0.5). (c) High-resolution XPS spectra of C 1s of Fe-MIL-101, xCu-Fe-MIL-101(CUMS) (x = 0.25, 0.5). (d) XPS valence band spectra. (e) NH3-TPD of Fe-MIL-101 and xCu-Fe-MIL-101(CUMS) (x = 0, 0.25, 0.5). (f) EIS curves of Fe-MIL-101 and xCu-Fe-MIL-101(CUMS) (x = 0, 0.25, 0.5).

Figure 3 (a) The relative viabilities of E. coli with the treatment of different concentrations of samples in the dark. (b) Antibacterial rate of E. coli treated with control, different samples (100 μg/mL) for 4 h in the dark. (c) Plate photographs of E. coli after treatments by 0.5Cu-Fe-MIL-101(CUMS) in the dark for different times and in the light for different times. (d, e) Antimicrobial rate of E. coli treated with 0.5Cu-Fe-MIL-101(CUMS) (100 μg/mL) in the dark and light for different times. (f, i) SEM images of untreated E. coli, subjected to Fe-MIL-101, Fe-MIL-101(CUMS), and 0.5Cu-Fe-MIL-101(CUMS) (250 μg/mL) in PBS buffer in the dark. Confocal images of (j) untreated E. coli, (k, m) E. coli treated with Fe-MIL-101, Fe-MIL-101(CUMS), and 0.5Cu-Fe-MIL-101(CUMS) (500 μg/mL) stained with Syto 9 and PI under 37 ℃ for 4 h in the dark. Results are presented as mean ± SD (n = 3). P<0.05, **P<0.01, ***P<0.001.

Figure 4 (a) Zeta potential of the samples. (b, c) UV–vis spectra and band gap energy of the samples. (d) DMPO spin-trapping EPR spectra of ·O2− for xCu-Fe-MIL-101(CUMS) (x = 0, 0.25, 0.5) in the dark and light. (e) DMPO spin trapping EPR spectra of ·O2− for 0.5Cu-Fe-MIL-101(CUMS) in darkness and different light times. (f) DMPO spin-trapping EPR spectra of ·OH for xCu-Fe-MIL-101(CUMS) (x = 0, 0.25, 0.5) in the dark and light. (g) DMPO spin trapping EPR spectra of ·OH on 0.5Cu-Fe-MIL-101(CUMS) in the dark and at different light times. (h) Photo-luminescence spectra of HPA for different samples processed in the dark. (i) Photoluminescence spectra of HPA treated 0.5Cu-Fe-MIL-101(CUMS) under dark and different light times.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
 下载:
下载: