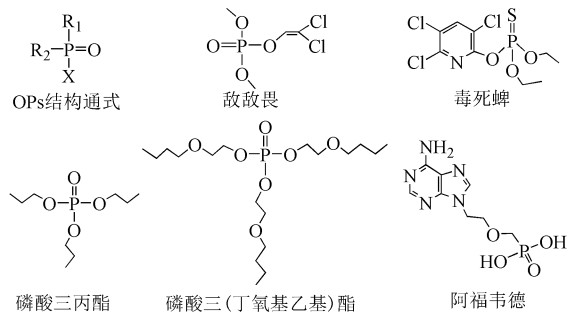
图式 1.
常见的有机磷化合物的结构式
Scheme 1.
The structures of common OPs

有机磷化合物(Organophosphorous compounds,OPs)广泛应用于阻燃剂[1, 2]、农药[3, 4]、抗病毒药物[5]、增塑剂、极压剂和抗磨剂中[6]。近年来,有机磷类阻燃剂作为溴代阻燃剂的替代品,已被广泛应用于工业化生产中,比较常见的有机磷阻燃剂有磷酸三(2-氯)异丙酯(TCIPP)、磷酸三(2-氯)乙酯(TCEP)等。OPs在农药中也有广泛使用,大多数为磷酸酯或硫代磷酸酯类化合物,比较常见的有敌敌畏、毒死蜱等。常见的OPs结构如图式 1所示,OPs结构通式中,R1、R2是烷基、烷氧基、烷硫基、芳香基;X作为离去基,通常是不稳定的酰基残基、卤化物、氰基、苯氧基或硫代物等,点表示键临界点。
伴随着OPs在各领域的应用,致使其在很多环境介质中被检测出来[7, 8],同时存在生物累积效应。许多OPs因具有抑制胆碱酯酶(ChE)活性导致胆碱酯酶失去水解乙酰胆碱(ACh)的能力而表现出毒性[9, 10]。OPs的毒性表现为急性毒性、迟发性神经毒性和细胞毒性,OPs中毒症状的表现取决于化合物的种类、剂量和中毒途径等[11]。OPs类物质有较高极性,可以在水中溶解,也可以经皮肤、呼吸道吸入,说明OPs可能会在生物体内富集并随生物迁移,存在生物累积效应。基于OPs的广泛应用,充分而快捷地了解OPs的毒性是非常必要的。
影响物质毒性的原因有很多种,例如结构与性质、机体状况、染毒条件和环境因素,物质的结构与性质对毒性的影响,目前的研究仍在发展阶段,本文主要讨论OPs的结构与性质对毒性的影响。根据毒理学知识,物质的结构与性质对活性的影响存在一些普遍规律:化合物结构中的卤素原子增多,导致分子极性增大,从而使化合物更易与酶系统结合,致使毒性增强;在芳香族化合物中引入羟基,毒性增强;脂/水分配系数高的化合物易于穿过细胞膜,使毒性增大。
通常是在体内或体外进行一些化合物毒性的试验来鉴定毒性程度和观察临床症状。由于OPs的高反应性毒性,使得实验危险因素高、不完整或数据难以获得。近年来,计算方法已经成为评估新的或未试验化合物的一种有效方法,可在选择药物、化妆品和农药方面提供一种低成本、时间少的高质量预测[12, 13]。相对于实验方法,计算方法的优势表现在绿色、快捷、便宜、准确以及可以对未合成出的化合物进行预测[14],因此构建高精度定量结构活性(QSAR)模型预测OPs的毒性是一个不错的方法。
定量结构性质/活性关系(QSPR/QSAR)是现在普遍应用的通过计算方法预测物质性质的一种理论研究方法,该方法在药物设计、材料化学、环境毒理学等领域都有广泛的应用。QSPR/QSAR方法是在经过理论计算之后,用统计方法对物质的描述符与其理化性质之间的相关性建模,进而揭示物质某种特征对性质的影响。Rybinska等[15]用遗传算法筛选描述符后用多元线性回归(MLR)建立的QSPR模型预测了离子液体的LogKow的值;Cao等[16]用QSAR方法结合量子化学描述符预测了离子液体的毒性;Paukku等[17]用QSAR方法研究了OPs的结构及其量子化学特征对毒性的影响;Edilson等[18]建立了肟、氨基肟和羟肟酸对有机磷制剂毒性抑制作用的QSPR模型,文中使用MLR方法建立的模型具有良好的验证参数,进而可以设计出抑制有机磷中毒的化合物。李钦玲等[19]用人工神经网络(ANN)预测了取代芳烃类化合物对黑呆头鱼的毒性,结果表明毒性与量化参数之间存在非线性关系。
本文中36个OPs的毒性数据收集于TOXNET (U.S. National Library of Medicine)网站的ChemIDplus数据库,本文采用大鼠急性口服毒性LD50(LD50是半数致死剂量,单位是mg/kg(体重))的对数,即logLD50表示毒性值(Aobs)。表 3列出的是36个OPs的结构以及毒性值等信息。
 下载:
导出CSV
下载:
导出CSV
| 序号 | 化学名 | 分子式 | CAS号 | Aobs | 结构 |
| 1 | Tris(o-ethylphenyl) phosphate | C24H27O4P | 3862-08-6 | 4.1987 |  |
| 2 | Tris(2-chloroethyl) phosphate | C6H12Cl3O4P | 115-96-8 | 3.0899 |  |
| 3 | Trioctyl phosphate | C24H51O4P | 78-42-2 | 4.5682 |  |
| 4 | O, O-Diethyl-S-(carbethoxy)methyl phosphorothiolate | C8H17O5PS | 2425-25-4 | 1.6532 |  |
| 5 | Pyridoxal phosphate | C8H10NO6P | 54-47-7 | 3.7709 |  |
| 6 | O, O-Dimethyl-S-2-(ethylsulfonyl)ethyl phosphorothioate | C6H15O5PS2 | 17040-19-6 | 1.5105 |  |
| 7 | Tris(1, 3-dichloro-2-propyl)phosphate | C9H15Cl6O4P | 13674-87-8 | 3.2672 |  |
| 8 | Tri-(2-chloroisopropyl)phosphate | C9H18Cl3O4P | 13674-84-5 | 3.1761 |  |
| 9 | Tri(3-chloropropyl) Phosphate | C9H18Cl3O4P | 26248-87-3 | 3.1761 |  |
| 10 | Tris(2, 3-dibromo-1-propyl) phosphate | C9H15Br6O4P | 126-72-7 | 2.9085 |  |
| 11 | O, O-Dimethyl-S-methylcarba- moylmethyl phosphorothioate | C5H12NO4PS | 1113-02-6 | 1.4771 |  |
| 12 | Tris(2, 4-xylenyl)phosphate | C24H27O4P | 3862-12-2 | 4.1987 |  |
| 13 | Triphenyl phosphate | C18H15O4P | 115-86-6 | 3.5441 |  |
| 14 | Tris(trimethylsilyl)phosphate | C9H27O4PSi3 | 10497-05-9 | 3.5366 |  |
| 15 | Tris(2-isopropylphenyl)phosphate | C27H33O4P | 64532-95-2 | 4.1987 |  |
| 16 | 7-[Bis(4-chlorobutoxy)phosphoryloxy]-3-chloro-4-methyl-chromen-2-one | C18H22Cl3O6P | 14745-61-0 | 2.0000 |  |
| 17 | 2, 3-Bis(diethoxyphosphinothioylthio)-1, 4-dioxane | C12H26O6P2S4 | 78-34-2 | 1.3010 |  |
| 18 | 5-Amino-1-[bis(dimethylamino)phosphinyl]-3-phenyl-1, 2, 4-triazole | C12H19N6OP | 1031-47-6 | 1.3010 |  |
| 19 | Tributyl phosphate | C12H27O4P | 126-73-8 | 3.4771 |  |
| 20 | Bis(2-chloroethyl) 2, 2-dichloroethenyl phosphate | C6H9Cl4O4P | 5266-08-0 | 1.3979 |  |
| 21 | Dibutyl phenyl phosphate | C14H23O4P | 2528-36-1 | 3.3304 |  |
| 22 | Dibutyl hydrogen phosphite | C8H19O3P | 1809-19-4 | 3.5051 |  |
| 23 | Phosphonic acid, ditridecyl ester | C26H55O3P | 36432-46-9 | 4.1761 |  |
| 24 | Phosphoric acid tris(4-aminophenyl) ester | C18H18N3O4P | 4232-84-2 | 2.1399 |  |
| 25 | Ethyl dihydrogen phosphate | C2H7O4P | 1623-14-9 | 3.2041 |  |
| 26 | Diisopropyl phosphite | C6H15O3P | 1809-20-7 | 3.2304 |  |
| 27 | Butanoic acid, 2, 2, 2-trichloro-1-(dimethoxyphosphinyl)ethyl ester | C8H14Cl3O5P | 126-22-7 | 3.0414 |  |
| 28 | Diethyl (2-chloroethyl)phosphonate | C6H14ClO3P | 10419-79-1 | 3.0000 |  |
| 29 | Diethyl hydrogen phosphite | C4H11O3P | 762-04-9 | 3.5911 |  |
| 30 | Bis(2-chloroethyl) 2-chloroethylphosphonate | C6H12Cl3O3P | 6294-34-4 | 2.9445 |  |
| 31 | Bis(2-ethylhexyl) hydrogen phosphite | C16H35O3P | 3658-48-8 | 4.0755 |  |
| 32 | Triethyl phosphate | C6H15O4P | 78-40-0 | 3.0663 |  |
| 33 | Trimethyl phosphate | C3H9O4P | 512-56-1 | 2.9243 |  |
| 34 | 3-Chloro-4-methylumbelliferonediethyl phosphate | C14H16ClO6P | 321-54-0 | 1.0000 |  |
| 35 | 2, 2, 2-Trichloroethyldihydrogen phosphate | C2H4Cl3O4P | 306-52-5 | 2.9294 |  |
| 36 | 3-Chloro-7-hydroxy-4-methylcoumarin bis(2-chloroethyl) phosphate | C14H14Cl3O6P | 321-55-1 | 2.9542 |  |
本文OPs的SMILES格式是从ChemIDplus搜索下载的,描述符的计算则是使用E-Dragon完成。E-Dragon是一个应用广泛的计算分子描述符的软件[20],所计算的分子描述符可以评价分子结构活性/性质的关系,以及用于分子数据库的相似性分析和高通量筛选,它可以计算分子的大量描述符。本文中,使用Dragon的在线版(E-Dragon at VCCLAB)[21],E-Dragon是Dragon的远程版本。Dragon提供了1600多种分子描述符,这些描述符被划分为20个逻辑块(结构描述符、拓扑描述符、行走和路径计数、连通性指标、几何描述符、电荷描述符、分子特性等),本文计算了20个逻辑块的所有描述符。
描述符通常根据维度来分类,例如结构描述符是直接从公式里推导出来(例如氧原子数),称为“零维”(0D)描述符;大多数属性描述符,例如n-辛醇/水分配系数或水的溶解度,被归类为“一维”(1D)描述符;拓扑描述符是基于分子图理论的“二维”(2D)描述符,描述了分子中单个原子之间的连接关系,第一个拓扑描述符在1947年由Wiener提出[22],此描述符相对简单没有考虑原子类型和键序,之后又有一些拓扑描述符被提出:Randic分子连接性指数χ[23]、Balaban指数J[24]、Kier和Hall指数[25]等;“三维”(3D)描述符表示的是在量子力学理论水平下优化三维分子结构的衍生属性;“四维”(4D)描述符表示由分子与周围空间的探针相互作用产生的分子特性或分子的立体动力学(包括键的灵活性、构像行为等)[26~29]。本文计算了Dragon所包含的所有描述符。
使用R语言对数据进行预处理,用R语言“caret”包nearZeroVar函数去除接近于常数的描述符。根据描述符与描述符两两之间的相关系数,使用findCorrelation函数去除相关系数大于0.9的描述符,筛选得到43个描述符,并使用R语言preProcess函数对数据中的描述符进行标准化,然后使用标准化后的数据进行后续的变量筛选及建模。使用R语言包建立线性核函数的支持向量机回归(SVM,由Cortes等[30]提出的一种算法)模型。本文用后退法作为变量筛选方法,以均方根误差(RMSE)作为评价标准对描述符进行筛选。
后退法是变量筛选的常用方法之一,其思想是先将所有的自变量都纳入模型进行计算,再逐一剔除自变量,直到没有可以剔除的自变量为止。在进行后退法计算的过程中,首先将所有变量都选入模型,生成m个回归系数对应的F检验值{F1,F2…,Fm},然后设min1={F1,F2…,Fm},在显著性水平α下,如果min1≤ Fα(1,n-m-1),剔除min1对应的自变量,记为x1。再将剩下的自变量纳入模型,建立回归方程,得到m-1个回归系数对应的F值。以此类推,直到得到最优的自变量子集,模型中的自变量即为最终确定的方程。
基于筛选出的14个描述符及36个OPs毒性数据,使用粒子群算法(PSO)来对参数Epsilon及惩罚因子C进行优化,PSO是通过群体中个体之间的协作和信息共享来寻找最优解。PSO中粒子所在位置的更新公式如下:
|
$ v_{i}^{d}=\omega v_{i}^{d}+c_{1} r_{1}\left(p_{i}^{d}-x_{i}^{d}\right)+c_{2} r_{2}\left(p_{g}^{d}-x_{i}^{d}\right) $ |
(1) |
|
$ x_{i}^{d}=x_{i}^{d}+\alpha v_{i}^{d} $ |
(2) |
其中,vid是粒子速度,ω是惯性因子,r1、r2是在0到1之间的随机数,xid是粒子当前位置。而c1与c2被称为加速常数,α被称为约束因子[31]。在优化过程中,设置Epsilon的取值范围为2-8至2-2(即0.003906至0.25),精度为0.01;惩罚因子C的取值范围为2-2至28(即0.25至256),精度同样设置为0.01,优化次数10000次。最终选择Epsilon=0.063,C=18.326,使用线性核函数的SVM算法建立预测OPs毒性的QSAR模型。
为了评价模型的准确度以及泛化能力,采用留一法(Leave-one-out,LOO)及外部数据验证方法对模型进行评价。对于回归模型,主要有3个评价指标:皮尔逊积矩相关系数R2、平均绝对误差MAE和RMSE,具体公式如下:
|
$ R^{2}=\left[\frac{\sum_{1}^{N}\left(x_{i}-\bar{x}\right)\left(y_{i}-\bar{y}\right)}{\sqrt{\sum_{1}^{N}\left(x_{i}-\bar{x}\right)^{2} \sum_{1}^{N}\left(y_{i}-\bar{y}\right)^{2}}}\right]^{2} $ |
(3) |
|
$ \mathrm{MAE}=\frac{\sum_{1}^{N}\left|x_{i}-y_{i}\right|}{N} $ |
(4) |
|
$ \operatorname{RMSE}=\sqrt{\frac{\sum_{1}^{N}\left(x_{i}-y_{i}\right)^{2}}{N}} $ |
(5) |
式(3)~(5)中,xi是试验值,yi是预测值,和是对应的平均值,N是样本数。
基于以上的计算结果,尝试使用ANN、MLR和偏最小二乘回归(PLS)建立预测OPs毒性的QSAR模型,并比较ANN、MLR、PLS和SVM这4个模型的优劣。ANN算法、MLR算法、PLS回归算法建立的QSAR模型同样使用R语言实现。
ANN是一种适用于模式识别和复杂的非线性函数关系拟合的方法[32],是模拟人体神经系统建立起来的信息系统,简单来说,神经网络是由具有适应性的单元组成的能够模拟生物神经系统对真实世界物体所作出交互反应的网络[33]。
MLR是QSAR常用的建模方法之一,MLR量化了不同的独立变量和因变量之间的关系。MLR广泛应用于预测磷酸酯毒性[34]。在本文模型中,用一组系数描述了自变量的线性组合,可以用下式将结构特征与毒性值联系起来:
|
$ \log L D_{50}=b_{0}+b_{1} x_{1}+b_{2} x_{2}+\cdots \cdots+b_{n} x_{n} $ |
(6) |
式(6)中,b0是截距,b1和b2是对应描述符的回归系数,x是描述化合物结构的描述符,n是这个方程中用来求出最优回归模型的描述符数量。MLR由描述符的逐步选择组成,包括向前和向后的过程。通常,最好的MLR模型应具有较高的预测能力和相关系数,并具有较低的标准误差。在MLR中,用复相关系数表示拟合程度的好坏。具体公式为:
|
$ R = \sqrt {1 - \frac{{\sum\limits_{i = 1}^n {{{\left( {{y_i} - {{\hat y}_i}} \right)}^2}} }}{{\sum\limits_{i = 1}^n {{{\left( {{y_i} - \bar y} \right)}^2}} }}} $ |
(7) |
式(7)中,yi为实验值,
PLS是20世纪70年代发展起来的基于多元回归、主成分分析(PCA)以及典型相关分析(CCA)的可以用来解决回归分析中自变量多重共线性问题的方法[35]。PLS可以排除原始变量的相关性,它既能过滤自变量的噪声,也能过滤因变量的噪声。此外,相比于主成分分析(PCA),PLS描述模型所需要的特征变量比PCA少,且预测能力也更加稳定。
使用E-Dragon at VCCLAB对36个OPs的20个逻辑块的描述符进行计算,共得到了1666个描述符:(a)48个结构描述符;(b)14个电荷描述符;(c)31个分子特征;(d)119个拓扑描述符;(e)47个行走和路径计数;(f)33个连通性指数;(g)47个信息指数;(h)96个自相关指数;(i)107个边界连接指数;(j)64个Burden描述符(BCUTs特征值);(k)21个拓扑电荷指数;(l)44个基于特征值指数;(m)41个Randic分子图像;(n)74个几何描述符;(o)150个径向分布函数;(p)160个基于电子衍射描述符结构的三维分子;(q)99个加权整体不变描述符(WHIMs) [36~38];(r)197个几何、拓扑和原子重量组合描述符(GETAWAY)[39, 40];(s)154种官能团的计数;(t)120个以原子为中心的片段。
本文使用R语言对数据进行预处理,用R语言“caret”包的nearZeroVar函数去除接近于常数的描述符。根据描述符与描述符两两之间的相关系数,使用findCorrelation函数去除相关系数大于0.9的描述符,最终得到43个描述符。并使用R语言preProcess函数对数据中的描述符进行标准化。同样使用R语言建立线性核函数的SVM模型。本文用后退法作为变量筛选方法,以RMSE作为评价标准对预处理后的43个描述符进行筛选,最终得到14个对SVM模型贡献较大的描述符(表 1)。其中5个是结构描述符(Me,Mp,RBF,nDB,nO),8个拓扑描述符(TI1,TIE,PW4,PJI2,T.N..O.,T.O..S.,T.O..P.,T.P..Cl.)和一个连接性指数(X5A)。文中用“caret”包对SVM模型筛选的描述符做了重要性分析,计算的重要性评分结果如表 1,文中对重要性评分大于0.2的6个描述符进行了简单解释。
下载:
导出CSV
| 描述符名称 | 描述符类别 | 描述符表达的意义 | 重要性评分 |
| Me[26] | 组成描述符 | 桑德森平均原子电负性 | 0.43161 |
| Mp[26] | 平均原子极化率(按碳原子比例) | 0.21460 | |
| RBF[26] | 可旋转键分数 | 0.06515 | |
| nDB[26] | 双键数 | 0.36825 | |
| nO[26] | 氧原子数 | 0.06621 | |
| TI1[41] | 拓扑描述符 | Mohar指数 | 0.42614 |
| TIE[42] | 电性拓扑参数 | 0.19173 | |
| PW4[43] | path/walk 4-Randic形状指数 | 0.12213 | |
| PJI2[44] | 二维Petitjean形状指数 | 0.30168 | |
| T.N..O.[45] | 氮原子和氧原子之间拓扑距离的和 | 0.03012 | |
| T.O..S.[45] | 硫原子和氧原子之间拓扑距离的和 | 0.15725 | |
| T.O..P.[45] | 磷原子和氧原子之间拓扑距离的和 | 0.23996 | |
| T.P..Cl.[45] | 磷原子和氯原子之间拓扑距离的和 | 0.04636 | |
| X5A[46] | 连通性指数 | chi-5平均连通性指数 | 0.05211 |
在本文的SVM模型中,计算的1666个描述符中Me对OPs毒性的影响评分最高,Me(mean atomic Sanderson electronegativity)表示桑德森平均原子电负性,即化合物的电负性等于各原子电负性的平均值[47]。同时本研究中Mp(mean atomic polarizability,平均原子极化率)对OPs毒性也有较大影响,与分子极化率对有机磷酸酯类化合物对发光菌的急性毒性有重要作用的研究结论相一致[48]。平均电负性Me和平均极化率Mp如式(8)和式(9):
|
$ M_{\mathrm{e}}=S_{\mathrm{e}} / n \mathrm{AT} $ |
(8) |
|
$ M_{\mathrm{p}}=S_{\mathrm{p}} / n \mathrm{AT} $ |
(9) |
式(8)和式(9)中,nAT是原子数量,Se是原子电负性之和,Sp是原子极化率之和,Se、Sp的计算如式(10)~(11)表示,e为电负性,p为极化率,i为第i个原子。
|
$ {S_{\rm{e}}} = \sum\limits_{i = 1}^A {{e_i}} $ |
(10) |
|
$ {S_{\rm{p}}} = \sum\limits_{i = 1}^A {{p_i}} $ |
(11) |
根据毒理学知识得知,含电负性基团的化学分子毒性强;另外根据研究发现,如果结构中存在具有强吸电子效应的卤素原子,则此结构更易与酶系统结合,从而导致卤素对化合物的毒性有重要影响[49]。本文中原子电负性对毒性有重要影响的结论与以上研究相一致。
对于Mohar指数TI1,是由拉普拉斯矩阵导出的,公式如下:
|
$ \mathrm{TII}=2 \log \left(\frac{\mathrm{B}}{\mathrm{A}}\right) W^{*} $ |
(12) |
式(12)中,A是A×A对称矩阵的顶点数,B是键数,W*是quasi-Wiener指数[50]。
nDB是双键数,OPs中存在的双键包括P=O双键、P=S双键、C=O双键和环内共轭双键等,双键的存在对OPs的毒性有一定的影响。PJI2是2维Petitjean形状指数,第一个Petitjean形状指数是拓扑等距描述符,也称为图论形状系数I2,I2定义如式(13)所示,式(13)中R和D是从分子图像中获得的拓扑半径和拓扑直径,表明OPs的毒性与其大小有关。
|
$ I_{2}=\frac{D-R}{R} 0 \leqslant I_{2} \leqslant 1 $ |
(13) |
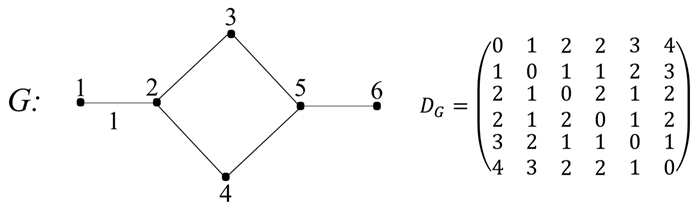
T.O..P.是磷原子和氧原子之间拓扑距离的和,对OPs的毒性重要性评分是0.23996,说明磷原子和氧原子之间拓扑距离的和对OPs的毒性有一定影响。研究发现,基于拓扑矩阵的拓扑指数与脂肪醇的毒性有关[51]。拓扑距离的和用距离矩阵D表示,具体算法如式(14),其中拓扑距离dij是原子顶点之间最短路径minPij的边数,此路径没有任何重复的顶点。图 2是一个图像及其距离矩阵示意图。
|
$ [D]_{i j}=\left\{\begin{array}{ll}{d_{i j}=\left|^{min}P_{i j}\right|} & {\text { if } i \neq j} \\ {0} & {\text { if } i=j}\end{array}\right\} $ |
(14) |
基于筛选出的14个描述符及36个OPs毒性数据,使用PSO来对参数Epsilon及惩罚因子C进行优化,设置Epsilon的取值范围为2-8至2-2(即0.003906至0.25),精度为0.01;惩罚因子C的取值范围为2-2至28(即0.25至256),精度同样设置为0.01,优化次数为10000次。最终选择Epsilon=0.063, C=18.326,使用线性核函数的SVM算法建立预测OPs毒性的QSAR模型。建模结果计算值与实际值的相关系数R为0.96,RMSE为0.268。
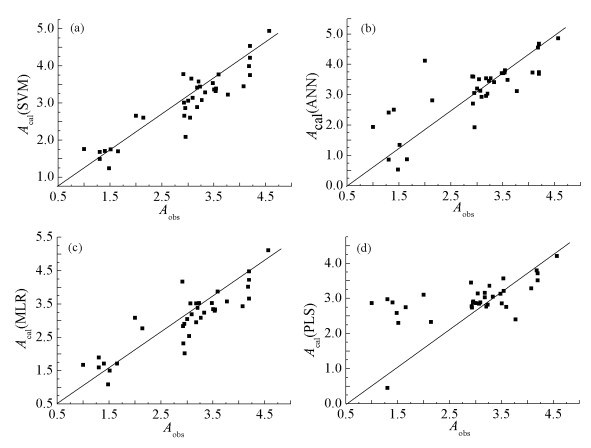
采用留一法及外部数据验证的方法对线性核函数的支持向量机QSAR模型进行评价。留一法结果显示,SVM预测OPs毒性的QSAR模型的计算值与实际值的相关系数R为0.913(如图 3(a)),RMSE是0.388。采用文献报道[34]的8个OPs进行外部数据验证,见表 4。结果显示,模型预测的平均相对误差为8.46%,说明该QSAR模型具有良好的泛化能力。
(a)SVM模型交叉验证结果;(b)ANN模型交叉验证结果;(c)MLR模型交叉验证结果;(d)PLS模型交叉验证结果
下载:
导出CSV
| 序号 | 化学名 | Aobs | 结构式 | Acal(SVM) | Acal(ANN) | Acal(PLS) | Acal(MLR) |
| 1 | Diisopropyl methyl phosphonate | 2.7900 |  |
2.3322 | 3.0116 | 2.9176 | 2.3687 |
| 2 | Bis(2-chloro-1-methylethyl)(2-chloro-1-methylethyl) phosphonate | 3.0000 |  |
3.1155 | 3.1219 | 3.1443 | 3.1801 |
| 3 | 1, 2, 2-Trimethylpropyl trimethylsilyl methyl phosphonate | 3.5000 |  |
3.5508 | 3.6768 | 3.2766 | 3.6342 |
| 4 | Diethyl [anilino(phenyl) methyl] phosphonate | 2.6000 |  |
2.7210 | 3.1036 | 2.9327 | 2.6700 |
| 5 | Diphenyl phenyl phosphonate | 3.3000 |  |
2.9912 | 3.3537 | 3.1810 | 2.9882 |
| 6 | 2-(Dimethoxy-phosphorylsulfanylmethyl)-5-methoxypyran-4-one | 1.2300 |  |
1.2375 | 0.8874 | 2.2771 | 1.2305 |
| 7 | 4-Bromo-2-chloro-1-[ethoxy(propyl-sulfanyl)phosphoryl]oxybenzene | 2.4700 |  |
2.2610 | 2.5760 | 3.0215 | 2.2666 |
| 8 | O, O-dimethyl-S-(2(ethylsulfinyl)ethyl)phosphorothioate | 1.6900 |  |
2.1642 | 1.7939 | 2.4033 | 1.9361 |
为了比较SVM模型与其他QSAR模型的优劣,采用ANN、MLR及PLS对筛选出的14个描述符进行建模,并用外部数据验证结果及留一法验证结果对模型进行评价。其中MLR模型如式(15)所示。
在使用ANN进行建模的过程中,设置输入层节点14个,隐蔽层节点8个,输出层节点1个,激活函数为sigmoid。训练集与测试集分别为36个与8个。图 3(b)、(c)、(d)和表 2显示,ANN建立的QSAR模型留一法验证结果相关系数R为0.686,RMSE为0.745;MLR模型建立的QSAR模型留一法验证结果相关系数R为0.878,RMSE为0.463;PLS建立的QSAR模型留一法验证结果相关系数R为0.620,RMSE为0.515。表 4列出了8个外部测试样本的结构、毒性值以及在不同算法下的预测值(Acal)。外部数据验证结果显示,ANN模型预测结果的平均相对误差为10.0%,MLR模型的QSAR模型预测结果的平均相对误差为7.00%,PLS的QSAR模型预测结果的平均相对误差为22.70%,综合比较四种算法的交叉验证结果、相关系数、RMSE和平均相对误差,发现ANN、MLR与PLS三种算法结果都劣于SVM预测结果。基于以上研究结果,最终选择线性核函数的SVM模型作为预测OPs毒性的QSAR模型。
下载:
导出CSV
| SVM | MLR | ANN | PLS | |
| 相关系数R(留一法) | 0.913 | 0.878 | 0.686 | 0.620 |
| 均方根误差(留一法) | 0.388 | 0.463 | 0.745 | 0.515 |
| 平均相对误差(外部测试集) | 9.10% | 7.00% | 10.0% | 22.7% |
|
$ A_{{\rm obs}}={\rm 9.255678(Me)-5.781683(Mp)-5.24027}\\{\rm (RBF)-1.248533(nDB)+0.2269104(nO)+}\\{\rm 0.008584594(TI1)+0.01185837(TIE)-9.703038}\\{\rm (PW4)+5.226757(PJI2)-0.02468373(T(N..O))}\\{\rm -0.0531226(T(O..S))+0.1058693(T(O..P))}\\{\rm -0.03822399(T(P..Cl))+14.19142(X5A)-6.38975} $ |
(15) |
本研究运用QSAR方法,结合E-Dragon,计算了36种OPs的1666个分子描述符。通过对描述符数据的预处理,去掉了相关性高和接近常量的描述符,共留下43个分子描述符。然后采用后退法对这43个描述符进行筛选,最终得到14个分子描述符。用线性核函数的SVM对筛选出的14个对OPs毒性有重要影响的描述符进行建模,然后对模型进行评价,同时对这14个描述符的重要性进行排序。另外,还建立了ANN、MLR以及PLS的OPs毒性预测模型,结果表明,用线性核函数的SVM交叉验证结果的相关系数、均方根误差、平均相对误差比ANN、MLR和PLS模型结果要好。基于以上研究结果,最终选择线性核函数的SVM模型作为预测OPs毒性的QSAR模型。相关描述符对OPs毒性影响的作用机制还需后续深入研究。
G W Wang, H Y Chen, Z K Du et al. Sci. Total Environ., 2017, 590~591(15):50~59.
Z K Du, G W Wang, S X Gao et al. Aquat. Toxicol., 2015, 161:25~32. doi: 10.1016/j.aquatox.2015.01.027
M Alfonso, R Duran, D Fajardo et al. Neurochem. Int., 2019, 124:130~140. doi: 10.1016/j.neuint.2019.01.001
A Derbalah, R Chidya, W Jadoon et al. J. Environ. Sci., 2019, 79:135~152. doi: 10.1016/j.jes.2018.11.019
D Clercq. Clin. Microbiol. Rev., 2003, 16:569~596. doi: 10.1128/CMR.16.4.569-596.2003
J Liu. Appl. Chem. Ind., 2018, 47(12):2706~2710.
D V Van, B J De. Chemosphere, 2012, 88(10):1119~1153. doi: 10.1016/j.chemosphere.2012.03.067
R Hou, Y P Xu, Z J Wang. Chemosphere, 2016, 153:78~90. doi: 10.1016/j.chemosphere.2016.03.003
T C Marrs. Pharmacol. Ther., 1993, 58(1):51~66.
K MacPhee-Quigley, P Taylor, S Taylor. J. Biol. Chem., 1985, 260(22):12185~12189.
H Sanderson, P Fauser, M Thomsen et al. J. Hazard. Mater., 2008, 154(1~3):846~857. doi: 10.1016/j.jhazmat.2007.10.117
R Naven, S Louise-May. Hum. Exp. Toxicol., 2015, 34:1304~1309. doi: 10.1177/0960327115605440
M H Baig, K Ahmad, S Roy et al. Curr. Pharm. Des., 2016, 22:572~581. doi: 10.2174/1381612822666151125000550
M D Segall, C Barber. Drug Discov. Today, 2014, 19(5):688~693. doi: 10.1016/j.drudis.2014.01.006
A Rybinska, A Sosnowska, M Grzonkowska et al. J. Hazard. Mater., 2016, 303:137~144. doi: 10.1016/j.jhazmat.2015.10.023
L D Cao, P Zhu, Y S Zhao et al. J. Hazard. Mater., 2018, 352(15):17~26.
Y Paukku, G Hill. Int. J. Quantum Chem., 2012, 112(5):1343~1352. doi: 10.1002/qua.22995
E B A Filho, A A Santos, B G Oliveira. J. Mol. Struct., 2017, 1133:338~347. doi: 10.1016/j.molstruc.2016.12.035
李钦玲, 杨玉良.计算机与应用化学. 2017, 34:5.
H Moriwaki, Y S Tian, N Kawashita et al. J. Cheminform., 2018, 10:4. doi: 10.1186/s13321-018-0258-y
I V Tetko, J Gasteiger, R Todeschini et al. J. Comput. Aid. Mol. Des., 2005, 19(6):453~63. doi: 10.1007/s10822-005-8694-y
H Wiener. J. Am. Chem. Soc., 1947, 69(1):17~20. doi: 10.1021/ja01193a005
M Randic. J. Am. Chem. Soc., 1975, 97(23):6609~6615. doi: 10.1021/ja00856a001
A T Balaban. SAR QSAR Environ. Res., 1998, 8(1~2):1~21. doi: 10.1080/10629369808033259
L H Hall, L B Kier. J. Chem. Inf. Comput. Sci., 35(6):1039~1045. doi: 10.1021/ci00028a014
R Todeschini, V Consonni. Handbook of molecular descriptors. Germany Weinheim:Wiley-VCH Verlag, 2000.
T I Opera, C L Waller, G R Marshall. J. Med. Chem., 1994, 37(14):2206~2215. doi: 10.1021/jm00040a013
M Ravi, A J Hopfinger, R E Hormann et al. J. Chem. Inf. Comput. Sci., 2001, 41(6):1587~1604. doi: 10.1021/ci010076u
O Mekenyan, S Dimitrov, P Schmieder et al. SAR QSAR Environ. Res., 2003, 14(5~6):361~371. doi: 10.1080/10629360310001623953
C Cortes, V Vapnik. Mach. Learn., 1995, 20(3):273~379.
A M Nassef, E T Sayed, H Rezk et al. Energ. Source. A, 2019, 41(17):2094~2103. doi: 10.1080/15567036.2018.1549171
P D Wasserman. Neural Computing Theory and Practice. van Nostrand-Reinhold, New York, 1989.
T Kohonen. Neural Networks, 1988, 1(1):3~16.
L Rosa, M Camacho, A T Eliazar et al. Mol. Divers., 2018, 22(2):269~280. doi: 10.1007/s11030-018-9819-2
S Wold, M Sjostrom. L Eriksson. Chemometr. Intell. Lab., 2001, 58(2):109~130. doi: 10.1016/S0169-7439(01)00155-1
R Todeschini, M Lasagni. J. Chemometr., 1994, 8(4):263~272. doi: 10.1002/cem.1180080405
R Todeschini, P Gramatica. Quant. Struct-Act. Rel., 1997, 16(2):113~119. doi: 10.1002/qsar.19970160203
R Todeschini, P Gramatica. Quant. Struct-Act Rel., 1997, 16(2):120~125. doi: 10.1002/qsar.19970160204
V Consonni, R Todeschini, M Pavan et al. J. Chem. Inf. Comput. Sci., 2002, 42(3):682~692. doi: 10.1021/ci015504a
V Consonni, R Todeschini, M Pavan et al. J. Chem. Inf. Comput. Sci., 2002, 42:693~705. doi: 10.1021/ci0155053
B Mohar. Stud. Phys. Theor. Chem., 1989, 63:1~8.
A Voelkel. Comput. Chem., 1994, 18:1~4.
M J Randic. J. Chem. Inf. Comput. Sci., 2001, 41:607~613. doi: 10.1021/ci0001031
M J Petitjean. J. Chem. Inf. Comput. Sci., 1992, 32:331~337. doi: 10.1021/ci00008a012
J Devillers, A T Balaban. Topological Indices and Related Descriptors in QSAR and Drug Design. Gordon & Breach, Amsterdam (The Netherlands), 2000.
L B Kier, L H Hall. Molecular Connectivity in Structure-Activity Analysis. RSP-Wiley, Chichetser (UK), 1986.
R T Sanderson. J. Chem. Edu., 1988, 65(3):227~231. doi: 10.1021/ed065p227
D Jiang, J G Zhou, N Li et al. Asian J. Ecotoxicol., 2014, 9(1):71~80.
须志平, 邵旭升.世界农药, 2010, 32(6):7~12. doi: 10.3969/j.issn.1009-6485.2010.06.002
S Markovic, I Gutman, Z Bancevic. J. Serb. Chem. Soc., 1995, 60:33~636.
任碧野, 许友, 陈国斌.化学学报, 1999, 57:563~571. doi: 10.3321/j.issn:0567-7351.1999.06.005

图 3 SVM、ANN、MLR、PLS模型留一法交叉验证结果实际值与预测值比较
Figure 3 The actual values are compared with the predicted values of the leave-one-out method for cross-validation of SVM, ANN, MLR and PLS model
(a)SVM模型交叉验证结果;(b)ANN模型交叉验证结果;(c)MLR模型交叉验证结果;(d)PLS模型交叉验证结果
表 3 36个有机磷化合物的毒性及结构列表
Table 3. Toxicity and structure of 36 organophosphorus compounds
| 序号 | 化学名 | 分子式 | CAS号 | Aobs | 结构 |
| 1 | Tris(o-ethylphenyl) phosphate | C24H27O4P | 3862-08-6 | 4.1987 | |
| 2 | Tris(2-chloroethyl) phosphate | C6H12Cl3O4P | 115-96-8 | 3.0899 | |
| 3 | Trioctyl phosphate | C24H51O4P | 78-42-2 | 4.5682 | |
| 4 | O, O-Diethyl-S-(carbethoxy)methyl phosphorothiolate | C8H17O5PS | 2425-25-4 | 1.6532 | |
| 5 | Pyridoxal phosphate | C8H10NO6P | 54-47-7 | 3.7709 | |
| 6 | O, O-Dimethyl-S-2-(ethylsulfonyl)ethyl phosphorothioate | C6H15O5PS2 | 17040-19-6 | 1.5105 | |
| 7 | Tris(1, 3-dichloro-2-propyl)phosphate | C9H15Cl6O4P | 13674-87-8 | 3.2672 | |
| 8 | Tri-(2-chloroisopropyl)phosphate | C9H18Cl3O4P | 13674-84-5 | 3.1761 | |
| 9 | Tri(3-chloropropyl) Phosphate | C9H18Cl3O4P | 26248-87-3 | 3.1761 | |
| 10 | Tris(2, 3-dibromo-1-propyl) phosphate | C9H15Br6O4P | 126-72-7 | 2.9085 | |
| 11 | O, O-Dimethyl-S-methylcarba- moylmethyl phosphorothioate | C5H12NO4PS | 1113-02-6 | 1.4771 | |
| 12 | Tris(2, 4-xylenyl)phosphate | C24H27O4P | 3862-12-2 | 4.1987 | |
| 13 | Triphenyl phosphate | C18H15O4P | 115-86-6 | 3.5441 | |
| 14 | Tris(trimethylsilyl)phosphate | C9H27O4PSi3 | 10497-05-9 | 3.5366 | |
| 15 | Tris(2-isopropylphenyl)phosphate | C27H33O4P | 64532-95-2 | 4.1987 | |
| 16 | 7-[Bis(4-chlorobutoxy)phosphoryloxy]-3-chloro-4-methyl-chromen-2-one | C18H22Cl3O6P | 14745-61-0 | 2.0000 | |
| 17 | 2, 3-Bis(diethoxyphosphinothioylthio)-1, 4-dioxane | C12H26O6P2S4 | 78-34-2 | 1.3010 | |
| 18 | 5-Amino-1-[bis(dimethylamino)phosphinyl]-3-phenyl-1, 2, 4-triazole | C12H19N6OP | 1031-47-6 | 1.3010 | |
| 19 | Tributyl phosphate | C12H27O4P | 126-73-8 | 3.4771 | |
| 20 | Bis(2-chloroethyl) 2, 2-dichloroethenyl phosphate | C6H9Cl4O4P | 5266-08-0 | 1.3979 | |
| 21 | Dibutyl phenyl phosphate | C14H23O4P | 2528-36-1 | 3.3304 | |
| 22 | Dibutyl hydrogen phosphite | C8H19O3P | 1809-19-4 | 3.5051 | |
| 23 | Phosphonic acid, ditridecyl ester | C26H55O3P | 36432-46-9 | 4.1761 | |
| 24 | Phosphoric acid tris(4-aminophenyl) ester | C18H18N3O4P | 4232-84-2 | 2.1399 | |
| 25 | Ethyl dihydrogen phosphate | C2H7O4P | 1623-14-9 | 3.2041 | |
| 26 | Diisopropyl phosphite | C6H15O3P | 1809-20-7 | 3.2304 | |
| 27 | Butanoic acid, 2, 2, 2-trichloro-1-(dimethoxyphosphinyl)ethyl ester | C8H14Cl3O5P | 126-22-7 | 3.0414 | |
| 28 | Diethyl (2-chloroethyl)phosphonate | C6H14ClO3P | 10419-79-1 | 3.0000 | |
| 29 | Diethyl hydrogen phosphite | C4H11O3P | 762-04-9 | 3.5911 | |
| 30 | Bis(2-chloroethyl) 2-chloroethylphosphonate | C6H12Cl3O3P | 6294-34-4 | 2.9445 | |
| 31 | Bis(2-ethylhexyl) hydrogen phosphite | C16H35O3P | 3658-48-8 | 4.0755 | |
| 32 | Triethyl phosphate | C6H15O4P | 78-40-0 | 3.0663 | |
| 33 | Trimethyl phosphate | C3H9O4P | 512-56-1 | 2.9243 | |
| 34 | 3-Chloro-4-methylumbelliferonediethyl phosphate | C14H16ClO6P | 321-54-0 | 1.0000 | |
| 35 | 2, 2, 2-Trichloroethyldihydrogen phosphate | C2H4Cl3O4P | 306-52-5 | 2.9294 | |
| 36 | 3-Chloro-7-hydroxy-4-methylcoumarin bis(2-chloroethyl) phosphate | C14H14Cl3O6P | 321-55-1 | 2.9542 | |
 下载: 导出CSV
下载: 导出CSV
表 1 筛选出的14个描述符及其意义
Table 1. The selected 14 descriptors and their meanings
| 描述符名称 | 描述符类别 | 描述符表达的意义 | 重要性评分 |
| Me[26] | 组成描述符 | 桑德森平均原子电负性 | 0.43161 |
| Mp[26] | 平均原子极化率(按碳原子比例) | 0.21460 | |
| RBF[26] | 可旋转键分数 | 0.06515 | |
| nDB[26] | 双键数 | 0.36825 | |
| nO[26] | 氧原子数 | 0.06621 | |
| TI1[41] | 拓扑描述符 | Mohar指数 | 0.42614 |
| TIE[42] | 电性拓扑参数 | 0.19173 | |
| PW4[43] | path/walk 4-Randic形状指数 | 0.12213 | |
| PJI2[44] | 二维Petitjean形状指数 | 0.30168 | |
| T.N..O.[45] | 氮原子和氧原子之间拓扑距离的和 | 0.03012 | |
| T.O..S.[45] | 硫原子和氧原子之间拓扑距离的和 | 0.15725 | |
| T.O..P.[45] | 磷原子和氧原子之间拓扑距离的和 | 0.23996 | |
| T.P..Cl.[45] | 磷原子和氯原子之间拓扑距离的和 | 0.04636 | |
| X5A[46] | 连通性指数 | chi-5平均连通性指数 | 0.05211 |
下载: 导出CSV
表 4 8个外部数据验证的毒性及结构列表
Table 4. Toxicity and structure of 8 external data validation
| 序号 | 化学名 | Aobs | 结构式 | Acal(SVM) | Acal(ANN) | Acal(PLS) | Acal(MLR) |
| 1 | Diisopropyl methyl phosphonate | 2.7900 | |
2.3322 | 3.0116 | 2.9176 | 2.3687 |
| 2 | Bis(2-chloro-1-methylethyl)(2-chloro-1-methylethyl) phosphonate | 3.0000 | |
3.1155 | 3.1219 | 3.1443 | 3.1801 |
| 3 | 1, 2, 2-Trimethylpropyl trimethylsilyl methyl phosphonate | 3.5000 | |
3.5508 | 3.6768 | 3.2766 | 3.6342 |
| 4 | Diethyl [anilino(phenyl) methyl] phosphonate | 2.6000 | |
2.7210 | 3.1036 | 2.9327 | 2.6700 |
| 5 | Diphenyl phenyl phosphonate | 3.3000 | |
2.9912 | 3.3537 | 3.1810 | 2.9882 |
| 6 | 2-(Dimethoxy-phosphorylsulfanylmethyl)-5-methoxypyran-4-one | 1.2300 | |
1.2375 | 0.8874 | 2.2771 | 1.2305 |
| 7 | 4-Bromo-2-chloro-1-[ethoxy(propyl-sulfanyl)phosphoryl]oxybenzene | 2.4700 | |
2.2610 | 2.5760 | 3.0215 | 2.2666 |
| 8 | O, O-dimethyl-S-(2(ethylsulfinyl)ethyl)phosphorothioate | 1.6900 | |
2.1642 | 1.7939 | 2.4033 | 1.9361 |
下载: 导出CSV
表 2 支持向量机与人工神经网络、多元线性回归、偏最小二乘回归模型比较结果
Table 2. SVM is compared with ANN, MLR and PLS
| SVM | MLR | ANN | PLS | |
| 相关系数R(留一法) | 0.913 | 0.878 | 0.686 | 0.620 |
| 均方根误差(留一法) | 0.388 | 0.463 | 0.745 | 0.515 |
| 平均相对误差(外部测试集) | 9.10% | 7.00% | 10.0% | 22.7% |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们


 下载:
下载: