Citation:
Sijia Zhang, Guangyu Bi, Wen-Qiang Li, Yang Wang, Xian-Yang Shi. Activated energy transfer pathway in carbon nitride nanosheets promotes molecular oxygen activation for robust and efficient water decontamination[J]. Chinese Chemical Letters,
2026, 37(4): 111463.
doi:
10.1016/j.cclet.2025.111463
Activated energy transfer pathway in carbon nitride nanosheets promotes molecular oxygen activation for robust and efficient water decontamination
English
Activated energy transfer pathway in carbon nitride nanosheets promotes molecular oxygen activation for robust and efficient water decontamination
School of Resources and Environmental Engineering, Anhui University, Hefei 230601, China
b.
CAS Key Laboratory of Urban Pollutant Conversion, Department of Environmental Science and Engineering, University of Science and Technology of China, Hefei 230026, China
Received Date:
02 January 2025 Accepted Date:
13 June 2025 Revised Date:
29 April 2025 Available Online:
15 April 2026
Abstract:
Carbon nitride (CN)-based photocatalytic processes hold promise for water decontamination, during which the widely acknowledged charge transfer pathway is unfortunately characterized by severe chemical stability challenges. Herein, the intensive yet underestimated energy transfer pathway is achieved in CN nanosheets for durable, efficient, and sustainable water purification. Upon simple oxygen activation in air, the synthesized nanosheets come with an optimal pollutant removal rate constant of 0.18 min−1 under visible light irradiation, far exceeding that of state-of-the-art photocatalysts. It is revealed that the enhanced exciton binding energy inside the CN nanosheets triggers strong exciton effects, promoting the accumulation of triplet-state excitons by regulating the energy gap. The accumulated triplet excitons almost accomplish the transition from oxygen exclusively to singlet oxygen via the energy transfer pathway, which ultimately contributes to the robust degradation of the pollutants. This work proposes a viable strategy to enhance the exciton effect within CN, upon which efficient water treatment technology driven by energy transfer pathways can be expected.
The global production and utilization of synthetic organic compounds (SOCs) have rapidly increased, with widespread applications in agriculture, industry, healthcare, and daily life [1,2]. These SOCs ultimately enter natural water bodies through industrial discharges, agricultural runoff, and urban sewage, posing significant challenges to environmental health and ecological safety [3]. Previous studies have shown that SOCs are prevalent in water bodies worldwide, frequently including dyes, antibiotics, and endocrine-disrupting chemicals (EDCs) [4–6]. These organic pollutants are known for their bioaccumulative and persistent nature, impacting water quality and potentially transferring through the food chain, ultimately posing risks to human health [7,8]. Due to its broad-spectrum capability, low energy consumption, and sustainability [9–11], sunlight-driven photocatalytic technology can potentially address this issue by removing SOCs. In this regard, the design and development of photocatalysts that balance low cost and high performance are key to the widespread application of photocatalytic water decontamination technology.
Carbon nitride (CN) is an organic conjugated polymer semiconductor with triazine (C3N3) or tri-s-triazine (C6N7) rings as the primary building blocks. Since the pioneering work reported in 2009, it has become a representative metal-free photocatalyst due to the combined merits of low production cost and flexible electronic structure [12]. With its inherent weakness reflected by the rapid recombination of photogenerated electrons and holes inside CN that severely limits its efficiency in photocatalytic decontamination [13], tremendous efforts have been devoted to enhancing the charge separation efficiency and improving the photocatalytic performance [14–16]. These endeavors contributed to the separation of photogenerated electrons and holes involved in the reduction and oxidative removal process of pollutants. It is important to note that efficient charge transfer processes are frequently accompanied by the production of strongly oxidizing reactive species [17,18]. These reactive species are prone to further attack the structure of CN, thereby posing stability concerns. Recent studies have revealed that strong oxidizing species (holes and hydroxyl radicals) derived from charge transfer processes inside CN can damage its structural motifs, leading to the self-decomposition of these photocatalysts [17–19]. Consequently, the CN-mediated photocatalytic decontamination system based on an effective charge transfer process is less likely to allow continuous operation and more prone to trigger secondary pollution.
On the other hand, the effect of excitons (electron-hole pairs) formed based on strong Coulomb interactions between photogenerated electrons and holes inside CN, has long been underappreciated in water treatment processes. These post-formation singlet excitons often undergo intersystem crossing (ISC) to transform into triplet excitons, which can activate oxygen (O2) to singlet oxygen (1O2) via energy transfer [20]. Such a pathway accompanied by selective 1O2 production can avoid the stability issue during the charge transfer process [21,22]. In this context, developing simple strategies to regulate the exciton effect and the ISC efficiency to boost the production of triplet excitons is highly desirable. Recent progress demonstrated that increasing the production of triplet excitons by regulating the thickness of two-dimensional (2D) materials is seemingly plausible, as reflected in the materials including graphene, black phosphorus, transition metal dichalcogenides. The weak dielectric screening and strong Coulomb interaction in the 2D materials significantly increase the exciton binding energy and stability, thereby evoking exciton effects [23–25]. Taken together, it is envisioned that reducing the thickness of CN increases the likelihood of inducing strong exciton effects, upon which the photocatalytic decontamination performance can be expected.
In this study, CN nanosheets with strong energy-transfer tendency, derived from the intense exciton effect, were prepared for proof-of-concept demonstration using a one-step heat treatment strategy. The morphological and structural characteristics of the target material, CN-650 nanosheet, were systematically investigated. Additionally, the superior decontamination performance of CN-650 nanosheets against SOCs, including dyes, antibiotics, and endocrine disruptors (EDCs) via O2 activation, was demonstrated. Particularly, 1O2, a key active species responsible for the robust and efficient degradation of pollutants through the energy transfer process, was identified and quantified. Furthermore, the underlying mechanism of the energy transfer pathway in the CN nanosheets, triggered by the strong exciton effect that mediates the exclusive conversion of O2 into 1O2, was carefully elucidated.
Melamine (C3H3N3, ≥99%), rhodamine B (RhB, ≥99%), bisphenol A (BPA, ≥99%), sulfamethazine (SMN, ≥99%), tetracycline (TC, ≥99%), humic acid (HA, ≥90%), sodium sulphate (Na2SO4, 99.5%), sodium nitrate (NaNO3, ≥99%), tert–butanol (t-BuOH, ≥99%), furfuryl alcohol (FFA, ≥98%), L-histidine (≥99%), p-benzoquinone (p-BQ, ≥99%), L-ascorbic acid (L-AA, ≥99%), nitrotetrazolium blue chloride (NBT, ≥99%), 5,5-dimethyl1-pyrroline-N-oxide (DMPO, ≥97%), and 2,2,6,6-tetramethyl-4-piperidone (TEMP, ≥99%) were purchased from Aladdin (Shanghai, China). Sodium chloride (NaCl, AR), sodium hydroxide (NaOH, AR), disodium ethylenediamintetraacetate (EDTA-2Na, AR), and hydrochloric acid (HCl, 37% AR) were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). Sodium dihydrogen phosphate (NaH2PO4, 99%), methyl orange (MO, 98%) were purchased from Macklin (Shanghai, China). All chemicals and reagents were used without further purification.
Briefly, 3 g of melamine precursor was placed in a quartz tube furnace and heated under a nitrogen atmosphere at a rate of 5℃/min to the target temperature (550, 600, or 650 ℃), held at the set temperature for 2 h. After cooling to room temperature, the powdered material was washed three times with deionized water and subsequently dried in a vacuum oven at 60 ℃ overnight. The as-synthesized CN samples were designated as CN-X, where X indicates the calcination temperature (550, 600, and 650 ℃).
The material characterization techniques are detailed in Supporting information.
Synthetic dyes fall into the category of SOCs, in which RhB as a typical representative was selected as the model pollutant due to its high-water solubility, non-volatility, and widespread use as a nitrogen-containing dye across various fields. The photocatalytic degradation of RhB was carried out in a 100 mL double-layer quartz jacketed beaker connected to a circulating condensate system, which maintained a reaction temperature of 25 ± 0.5 ℃. Visible light irradiation was provided by a 300 W xenon lamp equipped with a 420 nm cutoff filter. Specifically, 30 mg of photocatalysts were dispersed in 50 mL of 10 mg/L RhB solution. At each interval, 1.5 mL of the RhB solution was sampled and filtered through a mixed cellulose ester (MCE) membrane filter (pore size: 0.22 µm) to remove the solid species. The concentration of the samples was determined using a UV–vis spectrophotometer (UV–vis, X-6, YuanXi, China) at a detection wavelength of 554 nm. Additionally, various SOCs, including SMN, BPA, MO, and TC, were tested to further evaluate the photocatalytic performance of the prepared samples, following a procedure similar to that of RhB degradation. The spent photocatalysts were recollected by centrifugation at 15,000 rpm, washed with water, and dried at 80 ℃, which were then used for reusability and stability evaluation.
A glass sand-core filtration device was used as the reactor for continuous-flow experiments, with a polyvinylidene difluoride (PVDF) membrane (pore size: 0.22 µm) serving as the filter. A total of 100 mg of catalyst was loaded onto the PVDF membrane via vacuum filtration. The RhB solution (10 mg/L) was withdrawn from a beaker at a constant rate of 1.5 mL/min using a peristaltic pump. The other end of the filtration funnel was also connected to a peristaltic pump operating at the same rate to maintain a consistent filtration rate and stable liquid level. A xenon lamp was switched on at the beginning of the experiment and continuously illuminated the solution throughout the process.
Quenching experiments were conducted using t-BuOH to identify hydroxyl radicals (•OH), FFA, and L-histidine for 1O2, p-BQ for superoxide radicals (O2•−), and EDTA-2Na for photogenerated holes (h+). These experiments aimed to identify the reactive oxygen species (ROS) involved in the system [15,17,26].
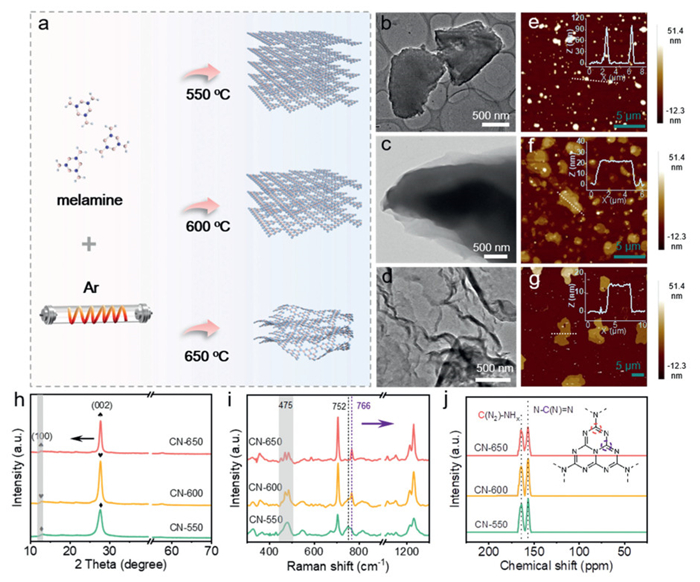
CN photocatalysts with different thicknesses were prepared by reasonably controlling the polymerization temperature, as schematically shown in Fig. 1a. The initial temperature of 550 ℃ was set to ensure a favorable condensation process [27] because lower temperature typically led to the formation of melem as a less-polymerized product (Fig. S1a in Supporting information). The highest temperature for polymerization was set as 650 ℃ based on the thermogravimetric results (Fig. S1b in Supporting information), which revealed the starting point of the fastest decomposition of CN. At this temperature, the CN samples achieved a yield of 42.7%, reflecting the balance between efficient polymerization and minimal decomposition. Once the temperature exceeded this threshold, massive structural defects that were not favorable for catalytic reactions were created, leading to a significant decrease in the material's quality and performance [28]. Field emission transmission electron microscopy (FE-TEM) results clearly show that the thickness of CN-X photocatalyst gradually decreases with increasing polymerization temperature (Figs. 1b-d). Higher temperature also came with a thermal exfoliation effect, leading to the concurrent thickness reduction and rolling up of these nanosheets. We further quantitatively determined the thickness of these samples using atomic force microscopy (AFM). As illustrated in Figs. 1e-g, the average thicknesses of CN-X were 92, 23, and 13 nm for CN-550, CN-600, and CN-650, respectively, again pointing to a reduction in thickness with increasing temperature.
Figure 1
Figure 1.
Schematic diagram of the preparation of CN-X photocatalysts (a). TEM images of CN-550 (b), CN-600 (c), and CN-650 (d). AFM images of CN-550 (e), CN-600 (f) and CN-650 (g), with insets showing height profiles of the corresponding samples. XRD patterns (h), Raman spectra (i) and solid-state 13C NMR spectra (j) of CN-X photocatalysts.
The crystalline structure evolution of CN-X photocatalysts with increasing temperature was carefully investigated by X-ray diffraction (XRD) patterns. The three samples shared similar diffraction signals (Fig. 1h), indicating that the crystalline phase is well preserved at high temperatures. Relative to that of the CN-550 sample, the strong diffraction peak of the (002) plane shifted towards a lower angle with increasing temperature, implying the enlarged interlayer distance accompanied by thickness reduction [29]. The peak at 12.8° assigned to the (100) plane gradually weakened with increasing temperature [30], signifying the less-ordered intra-plane packing of tri-s-triazine motifs due to the curved nanosheets, keeping consistent with the FE-TEM results [17,31,32]. The chemical environment and coordination structure of CN-X were probed by X-ray photoelectron spectroscopy (XPS), which revealed similar chemical environments of both C and N elements (Fig. S2 in Supporting information). The high-resolution C 1s XPS spectra of these samples can be divided into two peaks located near 288.16 eV and 284.80 eV (Fig. S2a), which belong to the C=N—C group in the aromatic heterocycle and the standard reference carbon, respectively [26,33,34]. The high-resolution spectra of N 1s comprise four signals located at 404.14, 401.26, 400.24, and 398.62 eV (Fig. S2b), corresponding to the π-π* bond in the aromatic heterocycle, -NH2, N-(C)3, and C=N—C of the tri-s-triazine ring structure, respectively [35,36]. Additionally, Fourier transform infrared (FTIR) spectroscopy results indicated that high temperatures did not significantly alter the functional group distribution of CN-X (Fig. S2), as all samples exhibited similar stretching and bending vibrations. The sharp peak at 810 cm−1 is assigned to the characteristic out-of-plane bending vibration of the tri-s-triazine ring sextant [37], consistent with the XRD results. A set of peaks centered at 1200–1750 cm−1 (Fig. S2c) are associated with the C—N and C=N stretching vibrations of the bridged or terminal amine groups in the tri-s-triazine structure [38]. The gradually intensified stretching signal at 3340 cm−1 (Fig. S2c) with increasing temperature is indicative of more -NH or -OH species on the CN surface [39,40]. The structural evolution of these photocatalysts was further investigated using Raman spectroscopy (Fig. 1i). Although they exhibit similar Raman shifts, the signal at 752 cm−1 assigned to the interlayer deformation vibration in CN-550 [41] shows a significant red shift in CN-600 and CN-650. This red shift can be attributed to the phonon confinement effect of the exfoliated nanosheets [30]. In addition, the characteristic peak of the tri-s-triazine ring located near 475 cm−1 splits into multiple peaks with increasing temperature [42], likely due to intraplanar curvature changes caused by the decrease in thickness. The results of 13C solid-state nuclear magnetic resonance (ss NMR) indicate that all CN-X samples present the same two signal peaks with chemical shifts of 156.6 and 164.3 ppm (Fig. 1j), attributed to the N—C(N)=N and C(N2)-NHx motifs in the tri-s-triazine structure, respectively [43,44]. Nitrogen sorption isotherms (Fig. S3 in Supporting information) reveal that the specific surface areas of CN-X increased with calcination temperature, which contributed to the exposure of active sites and adsorption of reactants [45]. Overall, the thickness of CN-X gradually decreased with increasing temperature while retaining the primary structural motif. Consequently, the sample CN-650, with the thinnest nanosheet was selected as the target material.
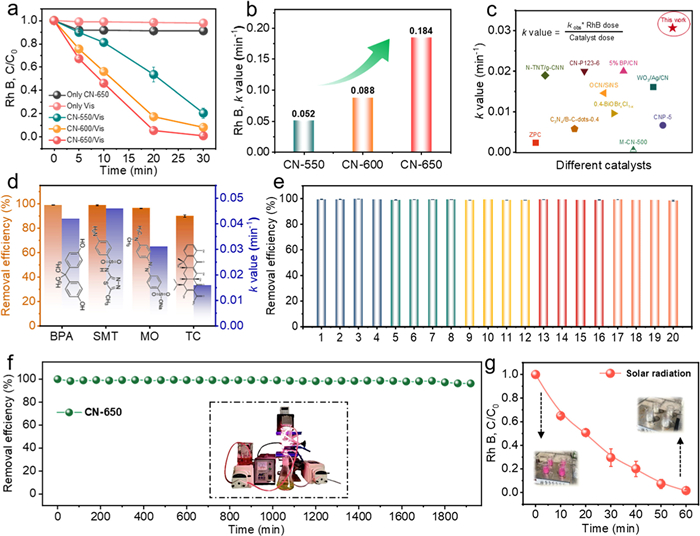
The photocatalytic activity of the prepared CN-X photocatalysts was investigated through RhB degradation experiments conducted in air under visible light irradiation. The results of the condition optimization experiments were shown in Fig. S4 (Supporting information), and the conditions ensured a balance between degradation efficiency and reaction kinetics [46]. As shown in Fig. 2a, the effects of adsorption in the dark and the self-decomposition of RhB induced by visible light were ruled out prior to the degradation experiments. Under visible light irradiation, CN-550, CN-600, and CN-650 removed 79.6%, 91.8%, and 100% of RhB within 30 min, respectively (Fig. 2a). The pseudo-first-order kinetic fitting results confirmed that the rate constant (k) for RhB degradation increased significantly with decreasing thickness of the CN samples, as indicated in Fig. 2b, CN-650 exhibited the highest k-value (0.184 min−1), which is 3.5 times greater than that of CN-550 (0.052 min−1) and superior to the state-of-the-art photocatalyst (Fig. 2c). Moreover, a comparative degradation experiment was conducted using representative metal oxides and sulfides, such as Fe2O3, Fe3O4, TiO2, and ZnS. The results demonstrated that CN-650 exhibited significantly superior degradation performance compared to these photocatalysts (Fig. S5 in Supporting information). Notably, CN-650 achieved outstanding photocatalytic activity without the use of any metal elements, underscoring its eco-friendly nature and superior catalytic efficiency. This metal-free design also eliminated the risk of secondary pollution from metal ion leaching, enhancing its potential for sustainable environmental applications. In addition, the performance of the CN-650/Air/Vis system under different pH conditions was explored by adjusting the initial pH of the RhB solution. Unlike the acidic conditions, which promoted the degradation process (Fig. S6a in Supporting information), the degradation efficiency of RhB gradually decreased with increasing pH (3–11). Nevertheless, CN-650 was able to completely remove 10 mg/L of RhB within 80 min under strong alkaline conditions (pH 11), demonstrating its favorable pH adaptability. CN-650 was also found to maintain high RhB removal efficiency in the presence of 20 mmol/L anions and 5 g/L natural organic matter (e.g., NO3−, Cl−, SO42−, H2PO4−, and HA), suggesting its considerable resistance to interfering species commonly found in wastewater environments (Fig. S6b in Supporting information). However, the presence of HCO3− exerted a certain inhibitory effect on the degradation efficiency of RhB. This is because HCO3− can easily react with ROS in the system, consuming the ROS and competing with RhB in the reaction [3,47]. Moreover, its hydrolysis raised the pH to about 9, and the resulting alkaline conditions further reduced RhB degradation efficiency. The good tolerance of the CN-650/Vis system was further confirmed through RhB degradation experiments conducted with different water qualities, including tap water and actual lake water (Fig. S6c in Supporting information). The slight decrease in the k-value may be attributed to the pH of the solutions. Encouraged by these results, we evaluated the efficiency of the CN-650/Air/Vis system for treating real dyeing wastewater (see Table S2 in Supporting information for the water quality parameters). As seen in Fig. S6d (Supporting information), CN-650 was capable of completely decolorizing the printing and dyeing wastewater within 100 min, demonstrating its potential for treating dye wastewater. Also, CN-650 has proven effective in eliminating other SOCs, such as antibiotics (e.g., SMN and TC) and EDCs (e.g., BPA), albeit coming with fluctuated rate constants that may relate to the intrinsic properties of these contaminants (Fig. 2d). The molecular structure of pollutants, particularly their core frameworks and substituent properties, critically influences degradation efficiency. These differences arise from the selective attack of singlet oxygen on specific functional groups and the distribution of electron-donating and electron-withdrawing substituents, which together govern degradation rates [48–50]
Figure 2
Figure 2.
(a) Degradation curves of RhB under different systems. (b) Rate constants of RhB degradation by various CN photocatalysts. (c) Comparison of rate constants for RhB degradation by different photocatalysts. (d) Performance of the CN-650/Air/Vis system for the removal of various pollutants. (e) Reusability test of CN-650/Air/Vis system. (f) Continuous-flow experiments mediated by CN-650, with an inset showing a homemade continuous-flow device. (g) Experiments on the removal of RhB by CN-650 under natural sunlight irradiation. Reaction conditions: Catalyst dosage = 0.6 g/L, [RhB] = [BPA] = [SMT] = [MO] = [TC] = 10 mg/L, pH 5, solar photodensity = 67.3 mW/cm2, photocatalyst loading capacity = 0.1 g, and flow velocity = 1.5 mL/min.
The reusability and stability of CN-650 were carefully evaluated through cycling experiments and characterizations of the spent photocatalyst. Remarkably, CN-650 retained nearly 100% removal efficiency of RhB after 20 consecutive cycles of batch testing (Fig. 2e), emphasizing its superior reusability. We also performed continuous-flow experiments toward RhB removal using a home-made circulating flow device (Fig. 2f inset) to further assess the robustness of CN-650. As shown in Fig. 2f, the removal efficiency of RhB did not obviously drop after continuous operation of the CN-650/Air/Vis system for 32 h. The XRD and FTIR results revealed that the spent CN-650 well maintained its crystalline structure and functional group distribution (Figs. S7a and b in Supporting information). These findings support the excellent structural stability of CN-650, which may be attributed to the avoidance of photo corrosion through the charge transfer pathway. Additionally, this stability minimized potential contamination issues, further enhancing its applicability. Under natural sunlight, CN-650 achieved complete RhB (10 mg/L) removal within 60 min (Fig. 2g).
In summary, the photocatalytic performance of CN materials was significantly enhanced with decreasing thickness, among which the optimal CN-650 holds promise for sustainable and cost-effective water purification in real water bodies.
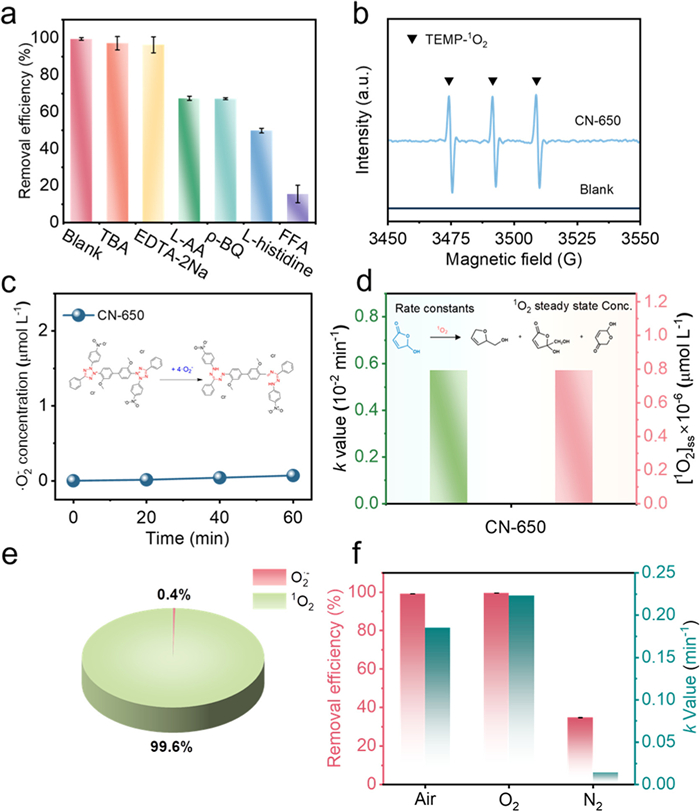
A series of quenching experiments were conducted to investigate the active species in the CN-X/Air/Vis system. In the CN-550 system, the presence of p-BQ significantly influenced the degradation efficiency of RhB (Fig. S8a in Supporting information), whereas the other two quenchers-TBA and FFA exhibited negligible effects. These findings suggest that O2•− was the primary reactive species responsible for RhB degradation. Notably, the addition of EDTA-2Na enhanced the degradation efficiency of RhB. It is possible since the oxidation of EDTA-2Na by photogenerated holes provides more photogenerated electrons involved in the generation of O2•− [51]. While in the CN-650 system, it is clear that the contributions of hydroxyl radicals (•OH) and holes (h+) to the degradation of RhB can be excluded, as the introduction of TBA and EDTA-2Na barely affected the process in Fig. 3a. In contrast, the degradation of RhB significantly inhibited when quenchers of O2•− (L-AA, 67.4% and p-BQ, 67.2%) and 1O2 species (L-histidine, 49.9% and FFA, 15.6%) were added, this holds particularly true for the latter (Fig. 3a). This suggests that these two types of ROS may play a significant role in the degradation process [52,53]. The above ROS were further qualitatively characterized using DMPO and TEMP as spin-trapping reagents and probed by electron paramagnetic resonance (EPR) spectroscopy [54,55]. DMPO-•OH signals (intensity ratio: 1:2:2:1) and DMPO—O2•− signals (intensity ratio: 1:1:1:1) were detected in the CN-550/Air/Vis system, while TEMP-1O2 signals were almost undetectable (Figs. S8b-d in Supporting information). This confirms that the primary ROS in the system was O2•−. Neither DMPO-•OH nor DMPO-O2•− signals were detected in the 650/Air/Vis system (Figs. S9a and b in Supporting information), implying that •OH and O2•− were not generated. This hints that the quenching effects of L-AA and p-BQ were more likely caused by false positives [56]. In contrast, a characteristic triplet signal (intensity ratio: 1:1:1) of the TEMPO product was detected in the system (Fig. 3b), which again demonstrates that 1O2 was the main ROS responsible for the efficient degradation of RhB [57]. The concentration of ROS was further determined using NBT and FFA as probes for O2•− and 1O2, respectively (Figs. S9c, S10a and S10b in Supporting information). The principles and implementation procedures for quantitative characterization are described in Supporting information [7,58,59]. The results in Fig. 3c show that there is almost no accumulation of O2•− in the system during the first 60 min, further excluding their contribution and aligning with the EPR results. The steady-state concentration of 1O2 was determined through the degradation experiment using FFA (Fig. 3d), revealing an actual concentration of ¹O2 species of 1.44 mmol/L based on Eqs. S2 and S3 (Supporting information). Subsequently, the contributions of the two ROS in the CN-650/Air/Vis system toward RhB degradation were ascertained, showing that the contribution of 1O2 was close to 100% (Fig. 3e). These results imply that CN-650 likely produces high-energy excitons via the exciton effect under visible irradiation, rather than utilizing photogenerated carriers through the charge transfer process. Besides, we performed RhB photodegradation experiments under different atmospheres to clarify the origin of 1O2 in the system. For the CN-550/Air/Vis system (Fig. S11 in Supporting information), the RhB degradation rate significantly decreased under nitrogen atmosphere, while oxygen introduction markedly enhanced the degradation efficiency. This phenomenon indicating that O2•− was most likely generated through the activation of oxygen molecules by photogenerated electrons [60]. For the CN-650/Air/Vis system, as evidenced in Fig. 3f, the introduction of O2 resulted in a 1.2-fold enhancement of the k-value, while a nitrogen atmosphere substantially suppressed this process, leading to an inhibition rate of 92.4%. These findings confirm that O2 in air is crucial for the photocatalytic generation of 1O2 by CN-650. Consequently, it can be inferred that CN-650 efficiently converted O2 from air into 1O2 species with nearly 100% selectivity via the exciton effect under visible light, ultimately facilitating the rapid elimination of RhB.
Figure 3
Figure 3.
(a) Efficiency of RhB degradation after the addition of different scavengers. (b) EPR spectra of TEMP-1O2 adducts. (c) Quantitative detection of O2•− in 650/Air/Vis system. (d) Kinetic rate constants of FFA and the corresponding steady-state 1O2 concentrations for 650/Air/Vis system. (e) Contribution of the two ROS in the 650/Air/Vis system. (f) Efficiency and kinetic constants of RhB degradation for CN-650 under different atmospheric conditions. Reaction conditions: [catalyst dosage] = 0.6 g/L; [RhB] = 10 mg/L; [TBA] = 500 mmol/L; [EDTA-2Na] = 10 mmol/L; [L-AA] = 10 mmol/L; [FFA] = [L-histidine] = 5 mmol/L; [p-BQ] = 2 mmol/L.
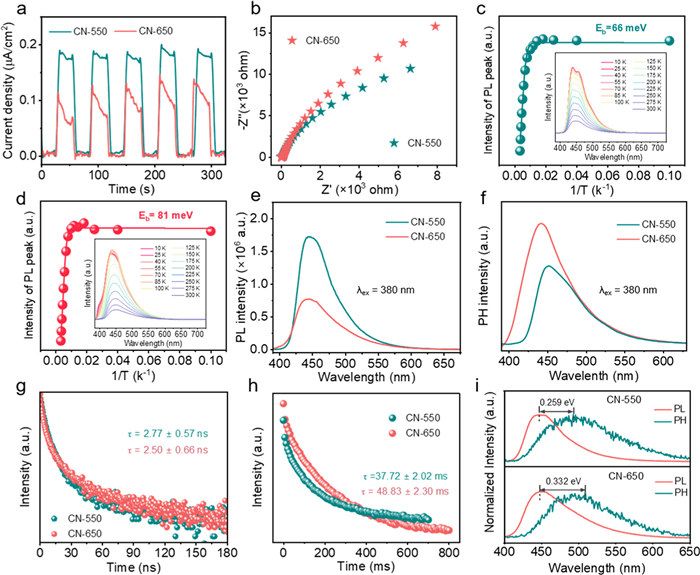
Next, extensive characterizations were conducted to elucidate the specific exciton pathway for the transformation of O2 to 1O2 in the CN-650/Air/Vis system. Firstly, the optical properties of CN-650 nanosheets were investigated using UV–vis diffuse reflection absorption spectroscopy. The results in Fig. S12a (Supporting information) show that the absorption edge of CN-650 nanosheets shifts from 460 nm to 420 nm compared to bulk CN-550, likely due to the exfoliation-induced quantum confinement effect resulting from thickness reduction [61]. Correspondingly, the band gap of CN-650 nanosheets increased from 2.82 eV to 3.02 eV, as determined from the Tauc plot (Fig. S12b in Supporting information), which is detrimental to the charge separation process [29,62]. The flat band positions of CN-550 and CN-650 derived from the Mott-Schottky plots (Figs. S12c and d in Supporting information) were 0.51 eV and 1.26 eV, respectively, and both are n-type semiconductors. Based on these results, the accurate energy band structures are obtained, as shown in Fig. S12e (Supporting information). The efficiency of the charge separation process within the CN-650 nanosheets was further evaluated through transient photocurrent curves and electrochemical impedance spectroscopy (EIS). Compared to bulk CN-550, CN-650 nanosheets exhibited both lower photocurrent (Fig. 4a) and higher charge transfer resistance (Fig. 4b), confirming the ineffective charge separation process within CN-650 nanosheets [63]. Real-time photocurrent monitoring reveal distinct RhB degradation mechanisms between CN-550 and CN-650 systems (Fig. S13 in Supporting information). The CN-650 system maintained a stable photocurrent, consistent with exciton-mediated energy transfer as the dominant degradation pathway. In contrast, CN-550 exhibited decreased photocurrent due to RhB adsorption blocking electron transfer, indicating distinct photocatalytic pathways. Furthermore, we calculated the exciton binding energies (Eb), which measure the strength of Coulombic interactions between photogenerated carriers. The value of Eb can be determined using variable temperature emission spectra, following the Arrhenius equation (Eq. 1):
Figure 4.
(a) Transient photocurrent response curve and (b) EIS Nyquist plots for CN-X. The intensities of integrated photoluminescence (PL) emission peak as a function of 1/T for CN-550 (c) and CN-650 (d). Insert: temperature-dependent emission spectra. (e) Steady-state PL and (f) phosphorescence (pH) spectra of CN-X. (g) The spectra of time-resolved PL (TR-PL) and (h) time-resolved pH (TR-pH) of CN-X. (i) Normalized PL and pH spectra of CN-X at 300 K.
where I0 represents the intensity of the photoluminescence (PL) peak at 0 K, kb is the Boltzmann constant, and T denotes the thermodynamic temperature (ranging from 10 K to 300 K) [58,64]. The results in Figs. 4c and d reveals larger Eb values for the CN-650 nanosheets (81 meV) compared to bulk CN-550 (66 meV), indicating a strong tendency toward exciton effects.
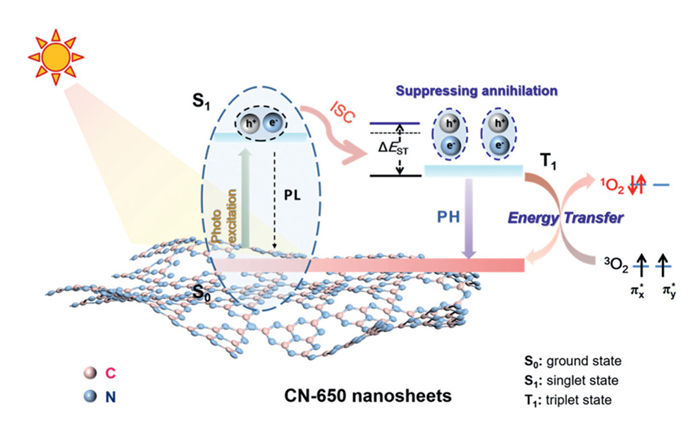
To gain insight into exciton effects in the CN-650/Air/Vis system, we performed steady-state and time-resolved PL and pH spectroscopic measurements. As illustrated in Fig. 4e, the PL intensity of CN-650 nanosheets is significantly quenched compared to that of bulk CN-550. This strong quenching implies that the transition from singlet excitons to triplet excitons occurs, rather than the separation of photocarriers, as supported by the reduced photocurrent and increased charge transfer resistance [20,65]. This observation is further corroborated by the pH spectra, where the pH intensity—representing the population of triplet excitons involved in the radiative decay process is substantially enhanced (Fig. 4f) [66]. These findings suggest that the reduction in thickness of CN enhances the conversion efficiency from singlet excitons to triplet excitons. The results of TR-PL and TR-pH spectroscopy provide dynamic insights into the exciton transition process. As depicted in Fig. 4g, the PL lifetime of CN-650 decreases from 2.77 ns to 2.50 ns, again supporting that the reduced thickness facilitates the efficient conversion of singlet exciton into triplet exciton [15,67]. Additionally, CN-650 with a pH lifetime of 48.83 ms is longer than that of bulk CN-550 (37.72 ms), indicating an extended lifetime of triplet exciton (Fig. 4h). Meanwhile, the energy gap (ΔEST) between the singlet and triplet states was estimated by normalizing the steady-state PL and pH spectra (Fig. 4i), where CN-650 nanosheets with a ΔEST value of 0.332 eV is higher than that of bulk CN-550 (0.259 eV). These results suggest that the increased energy gap suppresses nonradiative transitions within the CN-650 nanosheets, enhancing the lifetime and stability of triplet excitons by reducing annihilation among high-energy excitons, thereby increasing the yield of triplet excitons [68]. Femtosecond time-resolved transient absorption (TA) spectroscopy was employed to investigate the relaxation kinetics of the CN-X system. As shown in Figs. S14a and b (Supporting information), the longer lifetime observed in CN-550 indicates a slower rate of charge complexation, thereby providing photogenerated electrons with extended opportunities to participate in the reaction [69]. In contrast, CN-650 exhibited a comparatively shorter recovery lifetime for photoexcited excitons, facilitating the intersystem crossing (ISC) process required for triplet-state exciton generation [25]. These triple excitons play a crucial role in inducing the spin-flip transition of triplet oxygen (3O2) to form 1O2via energy transfer, ultimately leading to efficient pollutant removal (Fig. 5).
Figure 5
Figure 5.
Schematic diagram of energy level transitions inside CN-650.
This work demonstrates for the first time that the underestimated energy transfer pathway within carbon nitride is more conducive to allowing robust, efficient, and sustainable water purification processes compared to the well-known charge transfer pathway. Under visible light irradiation, the target photocatalyst CN-650 nanosheets exhibited considerably higher rate constants (0.184 min−1) for pollutant degradation than bulk CN-550 (0.052 min−1) and state-of-the-art photocatalysts in air conditions. The advantages of the CN-650/Air/Vis system are further highlighted by its extraordinary reusability and stability during the photocatalytic degradation of a wide range of SOCs, including dyes, antibiotics, and endocrine disruptors. Notably, the Eb value was considerably larger in the CN-650 nanosheets, inducing strong exciton effects. It was further demonstrated that the thickness inhibits the annihilation of triplet excitons by slightly increasing ΔEST, thereby enhancing the lifetime and population of triplet excitons. The accumulated triplet excitons facilitate the exclusive generation of 1O2 from O2 in the air via an energy transfer pathway, ultimately leading to efficient pollutant removal. This work provides a simple yet robust strategy to trigger energy transfer pathways within carbon nitride, holding promise for durable, efficient, and sustainable water decontamination processes.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figure 1
Schematic diagram of the preparation of CN-X photocatalysts (a). TEM images of CN-550 (b), CN-600 (c), and CN-650 (d). AFM images of CN-550 (e), CN-600 (f) and CN-650 (g), with insets showing height profiles of the corresponding samples. XRD patterns (h), Raman spectra (i) and solid-state 13C NMR spectra (j) of CN-X photocatalysts.
Figure 2
(a) Degradation curves of RhB under different systems. (b) Rate constants of RhB degradation by various CN photocatalysts. (c) Comparison of rate constants for RhB degradation by different photocatalysts. (d) Performance of the CN-650/Air/Vis system for the removal of various pollutants. (e) Reusability test of CN-650/Air/Vis system. (f) Continuous-flow experiments mediated by CN-650, with an inset showing a homemade continuous-flow device. (g) Experiments on the removal of RhB by CN-650 under natural sunlight irradiation. Reaction conditions: Catalyst dosage = 0.6 g/L, [RhB] = [BPA] = [SMT] = [MO] = [TC] = 10 mg/L, pH 5, solar photodensity = 67.3 mW/cm2, photocatalyst loading capacity = 0.1 g, and flow velocity = 1.5 mL/min.
Figure 3
(a) Efficiency of RhB degradation after the addition of different scavengers. (b) EPR spectra of TEMP-1O2 adducts. (c) Quantitative detection of O2•− in 650/Air/Vis system. (d) Kinetic rate constants of FFA and the corresponding steady-state 1O2 concentrations for 650/Air/Vis system. (e) Contribution of the two ROS in the 650/Air/Vis system. (f) Efficiency and kinetic constants of RhB degradation for CN-650 under different atmospheric conditions. Reaction conditions: [catalyst dosage] = 0.6 g/L; [RhB] = 10 mg/L; [TBA] = 500 mmol/L; [EDTA-2Na] = 10 mmol/L; [L-AA] = 10 mmol/L; [FFA] = [L-histidine] = 5 mmol/L; [p-BQ] = 2 mmol/L.
Figure 4
(a) Transient photocurrent response curve and (b) EIS Nyquist plots for CN-X. The intensities of integrated photoluminescence (PL) emission peak as a function of 1/T for CN-550 (c) and CN-650 (d). Insert: temperature-dependent emission spectra. (e) Steady-state PL and (f) phosphorescence (pH) spectra of CN-X. (g) The spectra of time-resolved PL (TR-PL) and (h) time-resolved pH (TR-pH) of CN-X. (i) Normalized PL and pH spectra of CN-X at 300 K.

 DownLoad:
DownLoad:

 下载:
下载:






 下载:
下载:
