Received Date:
28 August 2022 Accepted Date:
12 December 2022 Revised Date:
21 November 2022 Available Online:
15 August 2023
Abstract:
Small molecule activators could equally provide powerful tools as inhibitors do for interrogating cellular signal transduction. However, targeted protein activation is chemically challenging. Developing activators against Src homology region 2 domain-containing phosphatase-1 (SHP-1) to block STAT3 pathway represents a promising strategy for DLBCL therapy. Here we reported a new class of thieno[2,3-b]quinoline-procaine hybrid molecules as SHP-1 allosteric activators. The representative hybrid compound 3b displayed SHP-1 activating effect with EC50 of 5.48 ± 0.28 µmol/L. Further investigations confirmed that 3b allosterically interacted with SHP-1, switched it from close to open conformation, blocked SHP-1/p-STAT3 pathway, induced apoptosis and inhibited ABC-DLBCL cell proliferation in vitro, and delayed tumor growth in the xenograft model of SU-DHL-2. Overall, this work offered a novel paradigm to develop SHP-1 allosteric activators through chemical space evolution of PTPs inhibitors, and firstly validated the therapeutic strategy that directly activating SHP-1 alone could be a potential therapy against ABC-DLBCL via blocking STAT3 pathway.
The protein tyrosine phosphatases (PTPs) represent a large family of closely related enzymes regulating the homeostasis of protein tyrosine phosphorylation [1,2]. Malfunctions in PTPs activity are linked to various diseases, ranging from cancer to cardiovascular, immunological, infectious, metabolic, and neurological diseases [1,2]. Although all PTPs share a common mechanism for dephosphorylation, they lie in different locations and have different roles in signal transduction [3-7]. For example, protein tyrosine phosphatase 1B (PTP1B) works as a negative regulator involved in the insulin cascade and decreasing its activities has been regarded as a potential therapy for type 2 diabetes mellitus and obesity [8]. Meanwhile, Src homology-2 domain-containing protein tyrosine phosphatase 1 (SHP-1) is mainly restricted to hematopoietic and epithelial cells. It is widely accepted as a tumor suppressor via negatively regulating the cellular signaling processes [9], particularly controlling the signal transducer and activator of the transcription (STAT) pathway via dephosphorylate JAK kinases and STAT3 directly [10,11]. Activating SHP-1 may be another promising approach for anti-STAT3 cancer therapy. Hyperactivation of oncogenic STAT3 has been observed in diffuse large B-cell lymphoma (DLBCL), which is the most common type of non-Hodgkin's lymphomas with high invasiveness and heterogeneity [12]. Blocking STAT3 pathway is a potential therapeutic strategy for DLBCL [13]. Importantly, loss of function of SHP-1 promotes STAT3 deregulation in DLBCL [14]. SHP-1 is down-regulated in approximately 50% of primary DLBCL tumors [15,16]. Therefore, increasing SHP-1 activity represents a promising strategy for DLBCL therapy.
Enzyme activators are generally much more challenging to develop than enzyme inhibitors [17-19]. To our knowledge, nearly all of the reported compounds that can directly activate SHP-1 are kinase inhibitors including dovitinib [20-23], nintedanib [24,25], regorafenib [26], sorafenib and their derivatives (Fig. S1 in Supporting information) [27-34], and the numbers and structural diversities of SHP-1 activators are significantly less than that of PTP1B inhibitors developed over the past decades [35-39]. Therefore, imminent development of potent and specific SHP-1 activators with structural diversity remains necessary.
Our efforts to develop modulators of PTPs started from 1H-2,3-dihydroperimidines [40]. Using the scaffold hopping strategy, we developed 3-aryl-1-oxa-2, 8-diazaspiro[4.5]dec-2-enesbis-aryl amides [41] and 2-ethoxycarbonylthieno[2,3-b]quinolines as PTP1B inhibitors [42], benzo[c][1, 2, 5]thiadiazoles [43] and heterocyclic bis-aryl amides [44] as SH2-containing protein tyrosine phosphatase-2 (SHP-2) inhibitors, and 5-phenyl-1, 3, 4-thiadiazole derivatives as SHP-1 inhibitors (Fig. S2 in Supporting information) [45]. The high homology of PTP domain among PTPs makes it difficult to achieve inhibitor selectivity among closely related family members, meanwhile it opens a potential window to make switching between inhibitor and activator. Dushek group recently determined the distance that the closely related protein SHP-1 can reach from where it binds (SH2 domains) to where it exerts its catalytic activity (PTP domain), and found a value of 13 nm, to be compared with a distance of 5 nm in the crystal structure of the inactive state and of 20 nm if fully extended interdomain linkers are considered [46]. These results indicates that some inhibitors binding to catalytic subunit of homologic PTPs might have the chance to bind to SHP-1, disturb the conformational alignment between N-SH2 domain and PTP active site, and then switch it on. With this in mind, we screened our focused library of inhibitors of PTPs on SHP-1 biochemical assay and noticed that several thieno[2,3-b]quinoline derivatives showed activating effects against SHP-1 at the compound concentration of 50 µmol/L (Fig. S2). These results provided us a clue to explore novel small molecule activators of SHP-1. To construct a focused small molecule library, we introduced procaine and procainamide into the thieno[2,3-b]quinoline scaffold and identified compound 3b as a potent allosteric SHP-1 activator with higher selectivity than other PTP family members (Fig. S2). Mechanism studies indicated that 3b could allosterically activate SHP-1 through binding to N-SH2 domain. Furthermore, 3b inhibited tumor growth on human SU-DHL-2 cell xenografts in nude mice. Consequently, we offered a novel paradigm to develop SHP-1 activators through chemical space evolution of PTPs inhibitors, and firstly validate the therapeutic strategy that directly activating SHP-1 alone could be a potential therapy against ABC-DLBCL via blocking STAT3 pathway.
To identify a novel small molecule activator of SHP-1, we screened the in-house PTPs inhibitors library using 6, 8-difluoro-4-methylumbelliferyl phosphate (Difmup, Invitrogen D6567) as a substrate [47]. We noticed that thieno[2,3-b]quinoline derivatives with (4-(ethoxycarbonyl)phenyl)amino group (1a, 1b) and (4-methoxyphenyl)amino group (1c) showed apparent activating effects against SHP-1 at the concentration of 50 µmol/L. Meanwhile, the other types of groups, such as (4-acetylphenyl)amino group (1d), p-tolylamino group (1e), piperidine group (1f), (2-(diethylamino)ethyl)amino group (1g), and 4-(methoxycarbonyl)piperidin-1-yl group (1h), OEt (1i) and OH (2b), exhibited no effects against SHP-1 (Fig. S2). Based on the initial screening results, compounds 3a-3r were designed and synthesized by coupling thieno[2,3-b]quinoline-2-carboxylic acids (2a-2k) with procaine or procainamide (Scheme S1 in Supporting information), and compound 3b was hydrolyzed to obtain compound 3s (Scheme S1). To explore the effect of substituent at thiophene ring, compound 8 with amino group was prepared as outlined in Scheme S2 (Supporting information). Acetanilide 4 was treated with DMF and phosphorus oxychloride at 75 ℃ to obtain 5, which was condensed with hydroxylamine to afford oxime and subsequently treated with thionyl chloride to yield the 3-cyano-quinolone derivative 6. Without further purification, sulfhydrylation of 6 with thiourea afforded the compound 7. Then, compound 7 was reacted with 2-(diethylamino)ethyl 4-(2-bromoacetamido)benzoate under basic conditions in DMF to generate the target compound 8. To further examine the influence of thieno[2,3-b]quinoline scaffold on bioactivity against SHP-1, we fixed procaine as the optimal moiety, and 4-(quinolin-3-yl)thiophene-2-carbonyl group (compound 13), 2-(quinolin-2-ylthio)acetyl group (compound 16) and thieno[2,3-b]pyridine-2-carbonyl group (compound 18) were introduced. Synthesis of compounds 13, 16, 18 were outlined in Schemes S3-S5 (Supporting information) respectively. Compound 11 was produced via Suzuki coupling reaction. After saponification with lithium hydroxide in aqueous EtOH, compound 12 was obtained in 99% yield, followed by coupling with procaine to provide compound 13 in 18% yield (Scheme S3). Compound 15 was obtained in 98% yield by treating the bromide compound 14 with thiourea at reflux using absolute ethanol as solvent. After coupling compound 15 with 2-(diethylamino)ethyl 4-(2-bromoacetamido) benzoate, the compound 16 was yielded in 56% (Scheme S4). Compound 18 was prepared through amide coupling reaction (Scheme S5).
Among the compounds of 3a-3k with procaine scaffold, compounds 3b with a methyl group on position 8 showed the most efficient activating effect against SHP-1 (Table S1 in Supporting information). The activating effects were attenuated when methyl group on 8 position was replaced with other groups such as H (3a), OCH3 (3c), F (3d) and Br (3e). Transferring methyl group from 8 position to 9 position, the activating effect of compound (3f) dropped significantly. Among the compounds with methoxyl group (3c, 3g-3i), monosubstituted compound at 8 position (3c) displayed better activating effects than polysubstituted compounds (3h-3i), similar results were identified from compounds with methyl group (3b, 3f, 3j, 3k) and no compounds showed better activities than compound 3b. In general, compounds with procaine substitute (3a-3k) exhibited better activating effects than corresponding compounds with procainamide substitute (3l-3r). Introducing amino group into thiophene ring, Compound 8 showed lower activating effect against SHP-1 than corresponding compound 3a. These results indicated that the introduction of amino group on thiophene ring was detrimental to activating effect against SHP-1. Interestingly, Procaine and acid compound 3s hydrolyzed from 3b had no activating effect against SHP-1. Among compounds with procaine as the optimal moiety (13, 16 and 18), All the compounds dramatically decreased activating effects against SHP-1 compared with corresponding compound 3a (Table S2 in Supporting information). The results indicated that thieno[2,3-b]quinoline scaffold on compound 3a was necessary to maintain activating effect against SHP-1.
Overall, there are 125 human PTP superfamily genes [48], where only SHP-1 and SHP-2 contain two N-terminal SH2 domains. So here, full-length of SHP-2 was used to evaluate the selectivity of 3b. Other PTPs including PTP1B, TCPTP, only contain catalytic domain, which was used to reflect the effect of 3b on the catalytic domains of more other PTP families, except SHP-1 and SHP-2. As shown in Table S3 (Supporting information), compound 3b has no obvious activating effect against full length SHP-2, which shares a very similar active site and 61% amino acid homology in the sequence of SHP-1. Furthermore, 3b showed no effect on the PTP domain of SHP-1, SHP-2, PTP1B, TCPTP, and CDC25, confirming its high selectivity on PTPs catalytic domain. Previously reported SHP-1 activators including sorafenib [12] and their derivatives such as SC-43 [49] are kinase inhibitors. To make sure that compound 3b has good selectivity against kinases, several kinases related with cancer progress including BTK, LYN and JAK3 were selected (Table S4 in Supporting information). As expected, 3b showed poor inhibitory activities against selected kinases with IC50 of more than 30 µmol/L.
We then turned our attention to the biochemical properties of compound 3b (Fig. 1A) and noticed that compound 3b could directly activate recombinant human SHP-1 in a concentration-dependent manner (Fig. 1B). The different incubation times had no effect on the EC50 of compound 3b (Fig. 1C), indicating that compound 3b binds with SHP-1 in a fast-binding mode. To explore the activating mechanism of compound 3b against SHP-1, we measured the enzymatic kinetics of SHP-1 in the presence of compound 3b with the concentration of 50 µmol/L. The kinetic assays showed that compound 3b decreased the Michaelis constants (Km) by 1.98-fold and increased the turnover numbers (Kcat) by 8.31-fold. The data implied that the affinity of SHP-1 with substrate DiFUMP and catalytic efficiency were indeed enhanced by 3b (Fig. 1D). To further test the effect of 3b on biochemical dephosphorylation assay in the presence of activating p-Tyr peptide, 2P-IRS-1 peptide that could allosterically activate SHP-2 were used. Our biochemical assay showed that 2P-IRS-1 could also increase SHP-1 phosphatase activity by around 3.44 fold at 20 µmol/L (Fig. 1E), indicating 2P-IRS-1 could be the activating p-Tyr peptide of SHP-1. Furthermore, 3b still could increase SHP-1 phosphatase activity in the presence of 2P-IRS-1, showing as synergistic effect (Fig. 1F).
Figure 1
Figure 1.In vitro SHP-1 phosphatase assay for compound 3b in different conditions. (A) Structure of compound 3b. (B) Activation of full-length SHP-1 enzymatic activity by 3b, determined with DiFUMP as substrate. (C) The EC50 of compound 3b on SHP-1 in the different incubation times (0, 15, 30, and 60 min). 50 µmol/L of DiFUMP was added to start reaction. (D) Steady-state kinetic studies of SHP-1 activation with compound 3b. The initial velocities were measured at different concentrations of DiFUMP in the absence or presence of compound 3b at the concentration of 50 µmol/L. (E) Dose-dependent effects of 2P-IRS-1 on the activation of SHP-1 phosphatase. (F) Effects of 3b on SHP-1 phosphatase activation after treatment without or with 2P-IRS-1. Data are presented as mean ± S.D., n = 3, *P < 0.05, **P < 0.01.
Then we explored the possible interaction regions between SHP-1 and compound 3b by using a series truncated SHP-1 (Fig. S3A in Supporting information), including the PTP domain of SHP-1 (SHP-1-PTP Domain), SHP-1 without C tail (SHP-1-Delete C Tail), SHP-1 without N-SH2 domain (SHP-1-Delete N-SH2), and SHP-1 without C tail and N-SH2 domain (SHP-1-Delete N-SH2-C Tail). As shown in Fig. S3B (Supporting information), the SHP-1-PTP Domain and SHP-1-Delete N-SH2-C Tail showed significantly higher enhanced activity than SHP-1 WT (SHP-1 wild type), SHP-1-Delete C Tail, and SHP-1-Delete N-SH2. As shown in Fig. S3C (Supporting information), 3b could still activate SHP-1-Delete N-SH2 with EC50 of 17.27 ± 4.48 µmol/L, but the maximum activation ability decreased significantly with only 2.21-fold, compared to that in SHP-1 WT, indicating the N-SH2 domain was involved in 3b induced activation. Meanwhile, 3b could also activate SHP-1-Delete C Tail with EC50 of 21.86 ± 0.29 µmol/L and the maximum activation ability was similar to that in SHP-1 wild type, indicating the C-tail region took positive part in 3b induced activation. Importantly, 3b could not further activate SHP-1-PTP Domain or SHP-1-Delete N-SH2-C Tail, further confirming the importance of both of N-SH2 and C-terminal region in the 3b induced activation, suggesting that compound 3b might interact with either or both of the two regions as a potential allosteric activator.
Isothermal titration calorimetry (ITC) was adopted to measure the binding affinity of compound 3b with SHP-1. In the ITC assay, compound 3b showed an equilibrium dissociation constant (Kd) of 2.7 µmol/L (Fig. S4A in Supporting information). The measured thermodynamic binding parameters were −7.13 kJ/mol and −25.14 kJ/mol for enthalpy change (ΔH) and entropy change (−TΔS), respectively, suggesting a dramatic entropy change. In the small-angle X-ray scattering (SAXS) assay, the spatial remodeling of SHP-1 changed significantly, and the N-SH2 domain was away from the PTP catalytic region in an open conformation status after addition of compound 3b (Figs. S4B and C in Supporting information). These results indicated that the compound 3b indeed directly bind to SHP-1, and change the inactivated closed state to activated open conformation.
Unsuccessful to obtain a co-crystal structure of compound 3b-SHP-1 complex, molecular docking was used to predict the binding mode of 3b and SHP-1. The results from biochemical assay (Fig. S3C) and the SAXS assay (Figs. S4B and C) indicated the N-SH2 domain was the possible binding region. Accordingly, the potential binding site was assigned to the N-SH2 domain of SHP-1 (Fig. S5A in Supporting information) via SiteMap module implemented in Schrödinger and Fig. S5B (Supporting information) presented a predicted binding mode of compound 3b with SHP-1. Compound 3b could form two hydrogen bonds (H-bonds) with the N-SH2 domain of SHP-1, including an H-bond with the Gln87 through the tertiary amine of the procaine moiety, and an H-bond with the His51 through the amide group, which might be the major driving force for the binding. Hydrophobic interactions from the surrounding residues might also contribute to the binding, such as the hydrophobic side chains of Arg33, Lys34, His51 and Arg89. We also explored the selectivity of 3b towards SHP-1 and SHP-2. As shown in Fig. S5C (Supporting information), a lot of sequence differences could be found between the potential binding sites of two proteins. Fig. S5D and Fig. S5E (Supporting information) demonstrated the docking poses of 3b in SHP-1 and SHP-2 N-SH2 domains, respectively, which might also illustrate the difference between two pockets to some extent. The native groove formed by Arg33, Lys34, and Leu12 in SHP-1 could well fit the binding of the heterocycle moiety of 3b, while the corresponding Lys35, Ser36, and Val14 in SHP-2 could only result in a relatively flat surface that might be unfavorable for the binding of 3b.
To investigate the reason for the relatively higher activity of 3b, 3m was selected as a comparison and 50 ns MD simulations were conducted for each system. As shown in Fig. S6A (Supporting information), both systems can reach equilibrium after ~20 ns MD simulations, and hence the final 10 ns trajectories are chosen for further analysis. Here we emphatically discuss the differences of the H-bonds between the two systems, and several major H-bond interactions with their occupancies higher than 1% are summarized in Fig. S6C (Supporting information). As can be seen, the oxygen atom on the ester group of 3b can form a stable H-bond with GLN87, which occupies more than 50% of the time over the whole trajectory, while the corresponding H-bond between the oxygen atom on the amide group of 3m can just obtain an occupancy of 10.8%.
Hence, we infer that the stability of the H-bond formed between the ester/amide group and Gln87 may be the primary driving force for the activity gap between 3b and 3m. The representative conformations depicted as Fig. S6B (Supporting information) can further verify our inference. 3b can fluctuate in a relatively small region due to the strong H-bond interaction with Gln87, whereas 3m has significantly deviated away from its initial conformation, suggesting its poorer matching with the pocket. To further confirm the theoretical prediction, we built up the G87A mutant SHP-1 enzymatic assay, validated the binding affinity of 3b to mutant SHP-1, and the results demonstrated that the binding affinity was decreased obviously (Fig. S7 in Supporting information), indicating that the H-bond with Gln 87 takes pivotal rule for the potency of compound 3b.
To assess compound 3b binding to SHP-1 in the cell, the label-free biophysical assay of cellular thermal shift assay (CETSA), a broadly applicable method for measuring drug-target interaction, was conducted [50]. OCI-Ly10 and SU-DHL-2 DLBCL cells were treated with or without compound 3b for 2 h and then degraded cellular SHP-1 at different temperatures was detected by Western Blotting. As shown in Figs. 2A and B, the SHP-1 band obviously disappeared at 48 ℃ in cells treated with compound 3b, compared to the temperature of 50 ℃ in cells treated with DMSO. Furthermore, 3b could destabilize SHP-1 in dose dependent manner in SU-DHL-2 cell lysates (Fig. 2C). Taken together, our data suggest that 3b may influence the thermal stability shift of SHP-1, and affect SHP-1 function, which could be related to its inhibitory effects on cell growth, although further studies are needed to reveal the detailed mechanisms of SHP-1 destabilization.
Figure 2
Figure 2.
The cellular thermal shift assay for the binding of compound 3b to SHP-1 in cell lysates. Western blot image and the cellular thermal shift assay show that 3b (40 µmol/L) decreased the thermal stability of SHP-1 protein in OCI-Ly10 (A), and in SU-DHL-2 (B) cells. 3b decreased the thermal stability of SHP-1 protein at 49 ℃ in SU-DHL-2 cells in dose dependent manner (C). Experiments were performed in triplicate with biologically independent samples. P values were determined by Student's T-test in Prism 6.0; ns: not significant (P > 0.05), *P < 0.05, **P < 0.01, ***P < 0.001.
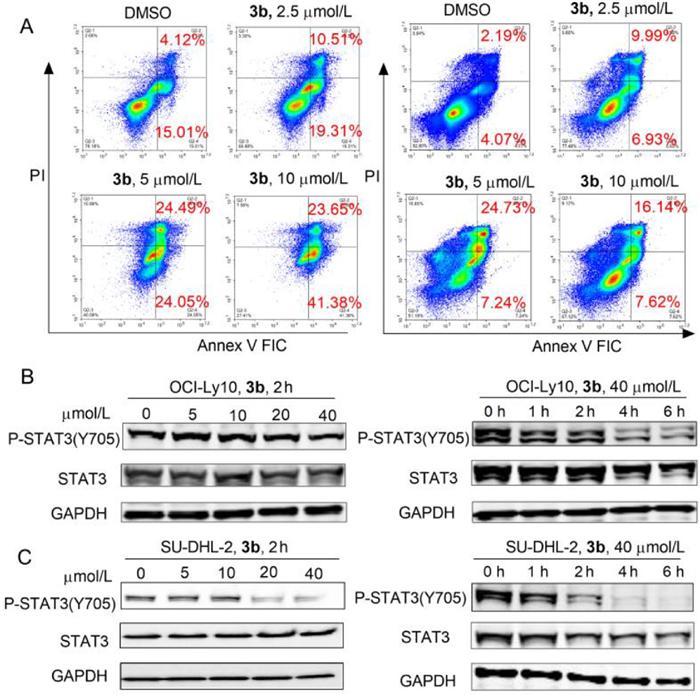
The compounds 1d, 1e, and 3b, 3j were chosen as negative and positive compounds respectively to evaluate their inhibitory activities against OCI-Ly10 cells and SU-DHL-2 cells (Table S5 in Supporting information). The negative compounds 1d and 1e showed no activities against OCI-Ly10 cells and SU-DHL-2 cells. Meanwhile, positive compounds 3b, and 3j inhibited the proliferation of OCI-Ly10 with IC50 of 2.73 ± 0.02 µmol/L, 2.86 ± 0.18 µmol/L, respectively, and only compound 3b showed inhibitory activities against SU-DHL-2 cells with IC50 of 8.39 ± 0.21 µmol/L, showing good correlation between the SHP-1 activation and cellular activity. The result indicated compound 3b could be used as a tool compound to validate the therapeutic strategy that directly activates SHP-1 to block the STAT3 pathway against DLBCL growth. Next, we used flow cytometry to detect cell apoptosis induced by compound 3b. The result revealed compound 3b increased the apoptosis of OCI-Ly10 and SU-DHL-2 cells in a dose-dependent manner (Fig. 3A). As SHP-1 could negatively regulate the phosphorylation level of tyrosine 705 of STAT3, we evaluated the effect of compound 3b on STAT3 in OCI-Ly10 and SU-DHL-2 cells by Western Blotting. The results showed that compound 3b reduced the phosphorylation of STAT3 in both OCI-Ly10 and SU-DHL-2 cells in a dose and time-dependent manner (Figs. 3B and C), indicating that compound 3b induced cell apoptosis via regulating the STAT3 pathway in ABC-DLBCL cells.
Figure 3
Figure 3.
Compound 3b induced cell apoptosis via downregulating the STAT3 pathway in DLBCL cells. (A) Compound 3b increased the apoptosis of OCI-Ly10 and SU-DHL-2 cells in a dose-dependent manner at 24 h. The apoptosis of cells was detected by flow cytometry with Annexin V/PI staining. Compound 3b inhibited the STAT3 pathway in both OCI-Ly10 (B) and SU-DHL-2 (C) cells in a dose and time-dependent manner. Western blot analyses were used to detect the effect of compound 3b on the phosphorylation of STAT3. Experiments were performed in triplicate with biologically independent samples.
We used the xenograft tumor model of SU-DHL-2 cells to evaluate the anti-DLBCL effect of compound 3bin vivo. The SU-DHL-2 cells were subcutaneously injected into the nude mice to establish the xenograft model. After the model was established successfully, we randomly grouped the mice into two groups. One group was intraperitoneally injected with 50 mg/kg compound 3b, and another group was intraperitoneally injected with the same volume of solvent as control. The treatment lasted 21 days, and the result showed that 50 mg kg−1 d−1 compound 3b significantly inhibited tumor growth (Figs. 4A and B) without noticeable body weight lost side effects (Fig. 4C) after 21 days of treatment. Animal projects were approved by The Institutional Animal Care and Use Committee (IACUC) of the Shanghai Institute of Materia Medica.
Figure 4
Figure 4.
Compound 3b inhibited DLBCL xenograft tumor growth in vivo. (A) The curve of tumor volume of the two groups of mice during the administration period were recorded. (B) The tumor weights of the two groups of mice after the administration were weighed. (C) The curves of body weight of the two groups of mice during the administration period were recorded. *P < 0.05.
The PTP superfamily typically share a conserved catalytic domain that catalyzes the enzymic reaction in which an active-site cysteine residue plays phosphatase activity. The close homology among members of PTP superfamily makes challenge to produce selective inhibitors against specific PTP, meanwhile it oppositely provides an opportunity to discover activators from related PTPs inhibitors. To date, known SHP-1 activators not only limit in the scant structures of pan-kinase inhibitors, but their molecular mechanisms also remain unclear, especially the biochemical and biophysical properties. The discovery of direct and selective SHP-1 activator still presents urgent challenges. In the present study, we identified a new class of SHP-1 activators, thieno[2,3-b]quinoline-procaine hybrid molecules, which evolved from PTP1B inhibitors. The representative hybrid compound 3b displayed SHP-1 activating effect with EC50 of 5.48 ± 0.28 µmol/L and high selectivity against SHP-1. Enzyme dynamic assay showed that compound 3b enhanced the affinity of SHP-1 to the substrate, as the Km value of the substrate decreased when the compound 3b was in the assay system. Moreover, we found that catalytic efficiency of enzyme also increased, as the Vmax and Kcat value also be increased by adding compound 3b in the assay system. In addition, biochemical truncation experiments indicated that compound 3b might interact with either or both of C tail and N-SH2 regions as a potential allosteric activator. Furthermore, using ITC and SAXS biophysical assay, we proved that compound 3b directly interacted with SHP-1 and switched SHP-1 from close to open conformation. Based on the results from biochemical and biophysical assays, the molecular docking predicted the binding mode of compound 3b with SHP-1 at the N-SH2 domain and explored the selectivity of 3b toward SHP-1 and SHP-2.
Although known SHP-1 activators kinase inhibitor including sorafenib and its derivatives have been used as tool compounds to investigate the function of SHP-1/p-STAT3 pathway in solid tumors such as liver, breast, and colon cancer [26-27,49]. They are seldom used to explore the importance of SHP-1 in non-Hodgkin's lymphomas. Therefore, we used compound 3b as a tool compound to elucidate the role of SHP-1 in ABC-DLBCL tumors and then identified that compound 3b inhibited the STAT3 signaling pathway to induce cell apoptosis by activating SHP-1. Notably, compound 3b inhibited ABC-DLBCL tumor growth in the SU-DHL-2 xenograft model. Taken together, we offered a novel paradigm to develop SHP-1 allosteric activators through the chemical space evolution of PTPs inhibitors and first validated a therapeutic strategy that directly activating SHP-1 alone could inhibit ABC-DLBCL via blocking the STAT3 pathway. Here, strong evidences including biochemical, Biophysical assay support 3b as the selective and direct SHP-1 activator, providing good starting point and reference for further development and exploration of SHP-1 activators. Further work will focus on optimizing the pharmacokinetic properties and investigating the binding mechanism of thieno[2,3-b]quinoline-procaine hybrid molecules with SHP-1 to develop specific activators to elucidate the functions of SHP-1 in DLBCL and other related diseases.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by National Natural Science Foundation of China (Nos. 81773779, 21772068 and 22277043), National Science & Technology Major Project "Key New Drug Creation and Manufacturing Program", China (No. 2018ZX09711002-007-1), Natural Science Foundation of Jiangsu Province (No. BK20190608) and Postgraduate Research & Practice Innovation Program of Jiangsu Province (No. KYCX22_2330). We also thank the staff from beamlines BL19U2 at Shanghai Synchrotron Radiation Facility (China) and the facility support from Bioduro-Sundia.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2022.108063.
[1]
R. Frankson, Z.H. Yu, Y. Bai, et al., Cancer Res. 77 (2017) 5701-5705.
[2]
A.J. Hale, E. Ter Steege, J. den Hertog, Dev. Biol. 428 (2017) 283-292.
A. Alonso, R. Pulido, FEBS J. 283 (2016) 1404-1429. doi: 10.1111/febs.13600
[49]
C.Y. Liu, L.M. Tseng, J.C. Su, et al., Breast Cancer Res. 15 (2013) R63.
[50]
D. Martinez Molina, R. Jafari, M. Ignatushchenko, et al., Science 341 (2013) 84-87. doi: 10.1126/science.1233606
Figure 1In vitro SHP-1 phosphatase assay for compound 3b in different conditions. (A) Structure of compound 3b. (B) Activation of full-length SHP-1 enzymatic activity by 3b, determined with DiFUMP as substrate. (C) The EC50 of compound 3b on SHP-1 in the different incubation times (0, 15, 30, and 60 min). 50 µmol/L of DiFUMP was added to start reaction. (D) Steady-state kinetic studies of SHP-1 activation with compound 3b. The initial velocities were measured at different concentrations of DiFUMP in the absence or presence of compound 3b at the concentration of 50 µmol/L. (E) Dose-dependent effects of 2P-IRS-1 on the activation of SHP-1 phosphatase. (F) Effects of 3b on SHP-1 phosphatase activation after treatment without or with 2P-IRS-1. Data are presented as mean ± S.D., n = 3, *P < 0.05, **P < 0.01.
Figure 2
The cellular thermal shift assay for the binding of compound 3b to SHP-1 in cell lysates. Western blot image and the cellular thermal shift assay show that 3b (40 µmol/L) decreased the thermal stability of SHP-1 protein in OCI-Ly10 (A), and in SU-DHL-2 (B) cells. 3b decreased the thermal stability of SHP-1 protein at 49 ℃ in SU-DHL-2 cells in dose dependent manner (C). Experiments were performed in triplicate with biologically independent samples. P values were determined by Student's T-test in Prism 6.0; ns: not significant (P > 0.05), *P < 0.05, **P < 0.01, ***P < 0.001.
Figure 3
Compound 3b induced cell apoptosis via downregulating the STAT3 pathway in DLBCL cells. (A) Compound 3b increased the apoptosis of OCI-Ly10 and SU-DHL-2 cells in a dose-dependent manner at 24 h. The apoptosis of cells was detected by flow cytometry with Annexin V/PI staining. Compound 3b inhibited the STAT3 pathway in both OCI-Ly10 (B) and SU-DHL-2 (C) cells in a dose and time-dependent manner. Western blot analyses were used to detect the effect of compound 3b on the phosphorylation of STAT3. Experiments were performed in triplicate with biologically independent samples.
Figure 4
Compound 3b inhibited DLBCL xenograft tumor growth in vivo. (A) The curve of tumor volume of the two groups of mice during the administration period were recorded. (B) The tumor weights of the two groups of mice after the administration were weighed. (C) The curves of body weight of the two groups of mice during the administration period were recorded. *P < 0.05.


 DownLoad:
DownLoad:



 下载:
下载:





 下载:
下载:
