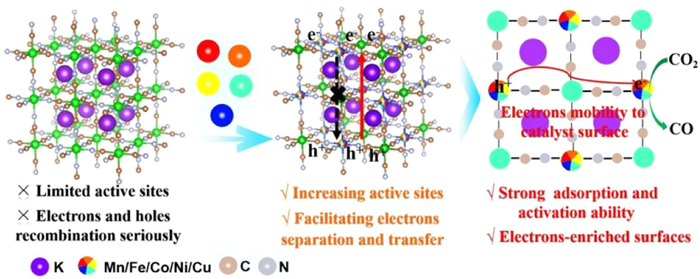
Figure 1.
Schematic diagram of reaction process. Schematic representation of CO2 activation and conversion to CO by HE-PBA.
Bioinspired high-entropy metal-organic frameworks towards boosted CO2 photoconversion
Xiaohong Li , Limin Jin , Haihui Yu , Yuteng Zhang , Haifeng Zhang , Yanbiao Liu

The excessive consumption of fossil fuels and associated emission of CO2 greenhouse gas have resulted in a severe environmental impact and energy crisis [1]. As highlighted by the National Academies of Sciences, Engineering, and Medicine, the removal of CO2 from atmosphere via chemical conversion into high-value chemicals or high-energy-density fuels is critical in achieving decarbonization, simultaneously addressing energy and environmental issues [2]. Solar radiation can serve as a renewable energy source to drive the CO2 reduction reaction (CO2RR), and researchers have been mimicking nature by employing light as a means of promoting chemical reactions over the past decades [3,4]. However, current advances in CO2RR are limited in terms of chemical thermodynamics and kinetics, due to a rapid charge recombination of electrons and "holes", the high C=O bond dissociation energy (~750 kJ/mol) and the requisite complex proton-coupled multi-electron reduction processes [5]. To overcome these obstacles, an appreciable research effort has been directed at developing advanced photocatalysts equipped with abundant active centers and effective charge separation characteristics [6-8].
In addition to the application of semiconductor/metal-oxide photocatalysts, the use of molecular photocatalysts is attracting significant attention. As it allows for precisely defined active sites and optimized electronic structure, potentially leading to desirable performance [9]. Of particular significance is the utilization of metal-organic frameworks (MOFs) assembled from metal ions/clusters with organic linkers, featuring well-defined crystalline structures, an intrinsic high porosity and tunable chemical compositions. These properties make MOFs ideal as platforms to explore structure-activity correlations in photocatalytic CO2 conversion [10-13]. Moreover, MOFs have been demonstrated to store large quantities of CO2 for extended periods, enabling application ranging from both carbon capture to conversion [14]. Notably, the metallic nodes in MOFs directly influence charge density and band structure, and can serve as active sites to bond CO2 molecules [15,16]. However, the majority of reported MOF systems suffer from intrinsic single metal sites and sluggish electron-hole separation, resulting in unsatisfactory performance in CO2 reduction [17,18].
Natural metalloenzymes bear multi-metallic catalytic complexes that promote the conversion of CO2 to CO. Researchers have designed mixed-metal MOFs to mimic metalloenzyme catalysts for CO2 reduction. Of particular relevance is the rational design of Ni-Fe-based carbon monoxide dehydrogenases (CODHs) where a NiI center binds to the carbon atom of CO2 whereas a FeII center binds to one oxygen atom, functioning as a Lewis acid to facilitate C-O bond cleavage. The two metals act synergistically to achieve a high selectivity in CO2 conversion [9,19,20]. In addition, Cu-Fe bimetallic photocatalytic MOFs have also exhibited enhanced CO2 conversion due to a synergistic effect involving Fe and Cu sites. In these systems, photo-induced electrons with a prolonged separated-state decay lifetime can be continuously generated [21]. A similar effect has been reported for Co-doped Zr-MOF and Mn-doped Zr-MOF in CO2RR [22,23]. Notably, researchers have also explored introducing metal Mn into MOF could induce the redistribution of local electron, due to its relatively lower electronegativity, metal Fe, Co and Ni would enhance the separation and capture of carrier [24,25]. In particular, metal Cu can facilitate transfer of electron to CO2. In this sense, the incorporation of dual metals (e.g., Mn, Fe, Co, Ni, or Cu) with varying electronegativity in MOFs results in distinct oxidative states, thereby functioning as donor and acceptor moieties. Such a catalyst design enhances overall redox activity and facilitates charge transfer within the framework that promotes photocatalysis [26,27]. The close proximity of donor and acceptor moieties enables a suitable alignment of electronic energy levels, accelerating charge localization via through-space or through-bond pathways [28]. The possibility of boosting activity by simultaneously assembling multi-metal sites in a high-entropy photocatalyst has yet to receive a comprehensive analysis and evaluation, which is the principal objective of this study.
Here, we propose a high-entropy (HE) strategy to promote CO2RR, incorporating five metals in Prussian blue (PB) as a type of MOF [24], to form HE-PBA. The catalytic active center should exhibit a tunable electronic structure and density, where a strong coupling due to the five constituent metals can induce a local redistribution of electrons that promotes the adsorption and activation of CO2. Light-driven charge carrier separation requires an interrelationship between electron donors (D) and acceptors (A). We have demonstrated that a combination of metal centers with different electronegativities facilitates effectively charge separation and transport. Notably, electron-enriched active sites can be formed by the induced migration of photo-generated electrons to the catalyst surface, thereby enhancing the reduction capability (Fig. 1). Taking Kx(MnFeCoNiCu)[Fe(CN)6] (HE-PBA) as an example, the material significantly enhances CO2 conversion, obtaining a CO yield of 1220.5 µmol g-1 h-1, which is appreciably higher than that achieved with medium-entropy samples based on water and CO2. The findings of this work highlight the potential of high-entropy materials in achieving superior CO2 photoreduction, offering a novel means of advanced CO2 treatment.
The Kx(MnFeCoNiCu)[Fe(CN)6] was prepared using a facile one-step hydrothermal method [24]. Also, preparation of the medium-entropy Kx(FeCoNiCu)[Fe(CN)6], Kx(MnCoNiCu)[Fe(CN)6], Kx(MnFeNiCu)[Fe(CN)6], Kx(MnFeCoCu)[Fe(CN)6], and Kx(MnFeCoNi)[Fe(CN)6] samples followed the same synthesis route, using the respective metal precursors. The metal ion content was determined by inductively coupled plasma-atomic emission spectrometry (ICP-AES). CO2 adsorption on the synthesized catalysts was evaluated in temperature-programmed desorption experiments (TPD) using a quartz reactor and thermal conductivity detector (TCD). Charge transfer was determined by X-ray absorption near-edge structure (XANES) spectroscopy. The direction of electron flow was measured by Kelvin Probe Force Microscopy (KPFM). Changes to the catalyst surface functional groups during CO2RR was probed by diffuse reflectance Fourier-transform infrared (FTIR) spectroscopy.
Details regarding catalyst characterization and performance evaluation are provided in Supporting information.
The HE-PBA was prepared in a one-pot hydrothermal synthesis using K4[Fe(CN)6] and equimolar quantities of divalent metal (Mn2+, Fe2+, Co2+, Ni2+ and Cu2+) precursors (Fig. 2a). The five cations (0.2 mol fractions) occupied the nitrogen-coordinated M positions, as illustrated in Fig. S1 (Supporting information). In addition, ME-PBAs samples with four equimolar cations (0.25 mol fraction) at the M position, serving as controls, were synthesized by removing one of the metal cations, i.e., Kx(FeCoNiCu)[Fe(CN)6] (ME-PBA(Mn)), Kx(MnCoNiCu)[Fe(CN)6] (ME-PBA(Fe)), Kx(MnFeNiCu)[Fe(CN)6] (ME-PBA(Co)), Kx(MnFeCoCu)[Fe(CN)6] (ME-PBA(Ni)), and Kx(MnFeCoNi)[Fe(CN)6] (ME-PBA(Cu)). The metal ion content was determined by inductively coupled plasma-atomic emission spectrometry (ICP-AES) (Table S1 in Supporting information). Powder X-ray diffraction (PXRD) analysis determined crystal structure, establishing a single-phase cubic Fm-3m space group (JCPDS #52–1907). The patterns exhibited peaks at 17.6°, 24.9°, 35.5°, 40.5°, 44.3°, and 50.6°, corresponding to (200), (220), (400), (420), (422), and (620) planes, respectively (Fig. 2b) [10]. Also, high-resolution transmission electron microscopy (HR-TEM, Fig. S2 in Supporting information) and aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC-HAADF-STEM) analysis have demonstrated a HE-PBA and ME-PBA interplanar spacing of 0.36 nm, corresponding to the (110) plane (Figs. 2c and d) [29,30].
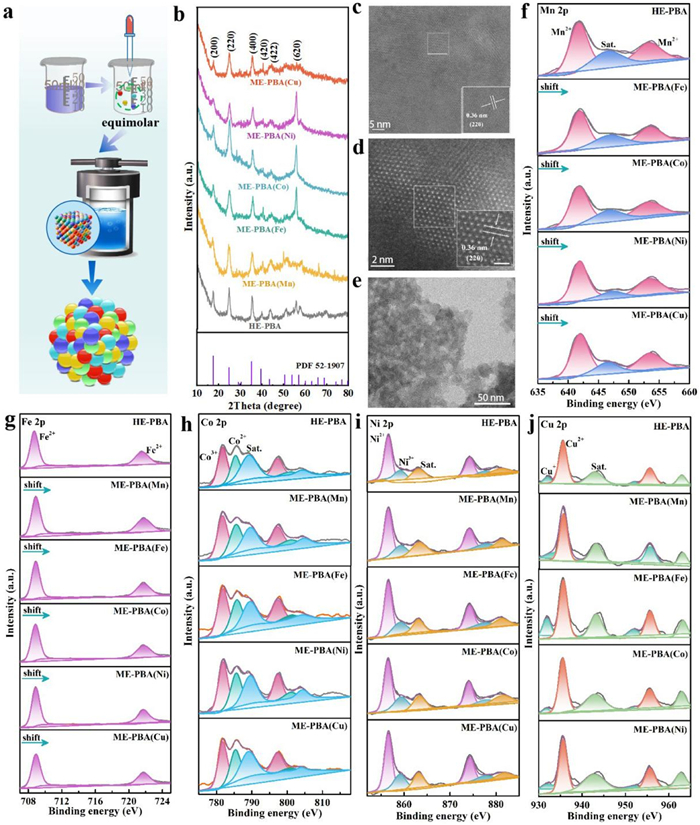
In the structural model, it is assumed that Fe1 atoms are coordinated to carbon at the 1a site, whereas the equimolar M (Mn, Fe2, Co, Ni and Cu) atoms coordinated to nitrogen share the 1b site. The lattice parameters for HE-PBA are a = b = c = 9.99 Å, α = β = γ = 90°, and V = 997.00 Å3. The structural model suggests the framework consisted of Fe1-C≡N-Mn/Fe2/Co/Ni/Cu-N≡C-Fe1 linear chains along the edges of the cubic unit cell, as shown in Fig. S1 [26,27]. To our delight, X-ray photoelectron spectroscopic (XPS) measurements have established the presence of five metal elements (Mn, Fe, Co, Ni and Cu) in the synthesized HE-PBA, whereas the ME-PBA samples were composed of four metal elements (with the exception of ME-PBA(Fe), Fig. S3a in Supporting information). In the case of both HE-PBA and ME-PBAs, three distinct carbon chemical states were observed with associated binding energies of 284.8 eV (C-C/C=C), 293.3 eV (C-O), and 296.2 eV (O-C-O) (Fig. S3b in Supporting information). The N 1s peaks were located at 397.9 eV (pyridinic nitrogen) and 402.0 eV (graphitic nitrogen) (Fig. S3c in Supporting information) [31,32]. The HE-PBA and ME-PBAs exhibited similar spherical nanoparticles with an average size of 20 ± 2 nm, as confirmed by scanning electron microscopy (SEM) and transmission electron microscopy (TEM) (Fig. 2e, Figs. S4 and S5 in Supporting information). Furthermore, high-angle annular dark-field scanning TEM (HAADF-STEM) and energy dispersive spectroscopy (EDS) elemental mapping of HE-PBA and selected ME-PBAs have clearly revealed a homogeneous distribution of metal ions without any detectable agglomeration, as illustrated in Figs. S6 and S7 (Supporting information). It should be noted that the Fe1 coordination with carbon resulted in a higher atomic mass fraction of Fe. The characterization measurements have established the successful synthesis of HE-PBA and a range of ME-PBAs.
The elemental composition and valence characteristics of the HE-PBA and ME-PBAs materials were further analyzed by XPS. In the case of HE-PBA (Fig. 2f), the high-resolution Mn 2p XPS spectra can be deconvoluted into three characteristic peaks corresponding to Mn2+ (641.6/653.1 eV) with a satellite peak at 646.5 eV [33,34]. The Fe 2p spectrum exhibited Fe 2p3/2 and Fe 2p1/2 peaks centered at 708.6 and 721.5 eV, corresponding to Fe2+ (Fig. 2g) [35]. The Mn 2p and Fe 2p associated with HE-PBA showed positive shifts relative to the ME-PBAs, indicating that Mn and Fe served as electron donors, facilitated by electronic interactions involving constituent Mn, Fe, Co, Ni, and Cu atoms. The XPS analysis established the presence of three Co species: Co2+ (785.3/800.8 eV), Co3+ (781.7/797.2 eV), and a satellite peak due to Co2+ (789.1/804.2 eV) (Fig. 2h) [36]. The Ni 2p spectra presented in Fig. 2i displayed primary peaks at 856.6 eV (Ni 2p3/2) and 874.2 eV (Ni 2p1/2), with satellite peaks at higher binding energies (863.2/880.9 eV), which are associated with Ni2+. Note that the minor peaks at 859.1 and 877.5 eV can be assigned to Ni3+ [37]. As for Cu 2p, a pair of peaks at 932.0/952.0 eV can be attributed to Cu+, whereas the peaks at 935.6 and 955.6 eV are due to Cu2+. In addition, the satellite peaks at 942.9 and 962.9 eV further confirm the presence of Cu2+ (Fig. 2j) [38]. The results have indicated an electron deficiency associated with Mn, Fe, Co, and Ni, and increased electron density for Cu, suggesting electron transfer from Mn, Fe, Co, and Ni to Cu atoms in HE-PBA. This transfer can be accounted for in terms of the differences in electronegativity (Mn < Fe < Co < Ni < Cu) [28]. The XPS results suggest strong interactions between the constituent metals that facilitate electron transfer.
The CO2RR photocatalytic activity of the synthesized catalysts was evaluated using a CH3CN/H2O and triethanolamine (TEOA) solution in the presence of Ru(bpy)3Cl2·6H2O (bpy = 2,2-bipyridine) and 1 atm CO2 at room temperature with a Xenon lamp irradiation for 1 h (λ > 420 nm). TEOA, as a sacrificial agent, is used to consume the photogenerated holes, while the photogenerated electrons participate in the reduction reaction. The liquid products, determined by 1H nuclear magnetic resonance (1H NMR), did not contain CH3OH, HCHO, or HCOOH (Fig. S8 in Supporting information). The photo-excited electrons generated following catalyst irradiation were transferred to CO2, and the consequent gas phase products were determined by gas chromatography (GC, Fig. S9 in Supporting information). To our delight, the HE-PBA outperformed ME-PBAs, delivering the highest CO production rate (1220.5 µmol g-1 h-1), accompanied by a H2 yield of 15.4 µmol g-1 h-1. In the case of ME-PBAs, the CO2RR activity varied significantly where ME-PBA(Cu) exhibited CO and H2 yields of 750.3 µmol g-1 h-1 and 7.1 µmol g-1 h-1, respectively and ME-PBA(Mn) generated a lower CO production (576.5 µmol g-1 h-1) with trace amounts of H2 (12.2 µmol g-1 h-1). By comparison, reaction over ME-PBA(Fe) and ME-PBA(Ni) resulted in CO production rates of 363.8 and 308.6 µmol g-1 h-1, respectively, with respective H2 evolution rates of 18.7 and 22.6 µmol g-1 h-1. In addition, CO2RR over ME-PBA(Co) resulted in the lowest activity of 261.4 µmol CO g-1 h-1 and 17.1 µmol H2 g-1 h-1, as shown in Figs. 3a and b, and Table S2 (Supporting information). The results have established superior CO2RR performance for HE-PBA, which can be attributed to a synergism associated with the constituent metals. In addition, the catalytic reduction was highly selective, producing CO as the predominant product with selectivity approaching 100% (Fig. 3c and Fig. S10 in Supporting information).
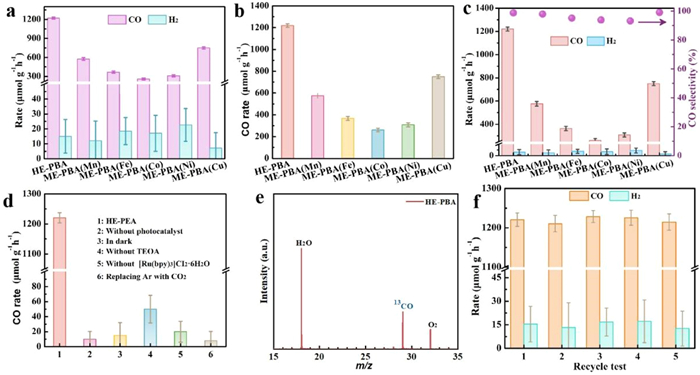
Control experiments presented in Fig. 3d and Table S3 (Supporting information) confirmed that no products were detected in the absence of catalyst, light irradiation, TEOA, Ru complex photosensitizer, or CO2, establishing the photocatalytic CO2RR using HE-PBA [28]. Isotope-labeled CO2 was screened to identify the carbon source of the observed products. The results have revealed a mass-charge ratio of 29 (m/z = 29) generated from 13CO2 that can be assigned to 13CO, confirming that the product was indeed derived from CO2 reduction (Fig. 3e) [39]. Moreover, the apparent quantum efficiency (AQE) of HE-PBA decreased with increasing irradiation wavelength (Fig. S11 in Supporting information), indicating that the photo-generated carriers were mainly responsible for CO production: the highest AQE was approximately 3.6% at 420 nm [40]. The HE-BPA are insoluble in the water and very stable during the photocatalytic testing experiments for CO2 reduction, which have good stability and durability, which is well proven in cyclic tests. The photostability of HE-PBA was examined over five consecutive cycles, and there was no discernible loss of activity (Fig. 3f). Moreover, there were no detectable changes in material morphology or crystalline following the cycling test (Fig. S12 in Supporting information), highlighting the robustness of the as-designed catalyst (The catalyst performance of HE-PBA was compared with other literature reports in Table S4 in Supporting information).
Temperature-programmed desorption (TPD) measurements were conducted to assess the CO2 adsorption sites. As shown in Figs. 4a and b, four stages of desorption occur at 119, 216, 298–368, and 443 ℃ for HE-PBA, ME-PBA(Mn), ME-PBA(Fe), ME-PBA(Co), and ME-PBA(Ni), which associated with the removal of chemisorbed CO2. In contrast, ME-PBA(Cu) displayed only two lower chemisorption peaks at 128 and 401 ℃, indicating fewer adsorption sites and a weaker CO2 chemisorption capacity [41]. The TPD results indicate that in the absence of Cu metal, the catalyst exhibits reduced chemical adsorption peaks and diminished CO2 adsorption capacity. This demonstrates that the Cu component functions as a critical adsorption site, effectively enhancing CO2 adsorption and thereby providing a solid foundation for facilitating subsequent catalytic reactions. Furthermore, the adsorption ability and BET surface areas of the catalysts were evaluated. The N2 adsorption-desorption isotherms exhibited a Type-Ⅳ profile with distinct hysteresis loops at high relative pressures (P/P0), confirming the presence of a mesoporous structure. The clear hysteresis loop indicates strong interaction at the binding site (Fig. S13 in Supporting information). Notably, the BET surface area of the catalysts remained largely unchanged (Table S5 in Supporting information), indicating structural stability under the tested conditions.
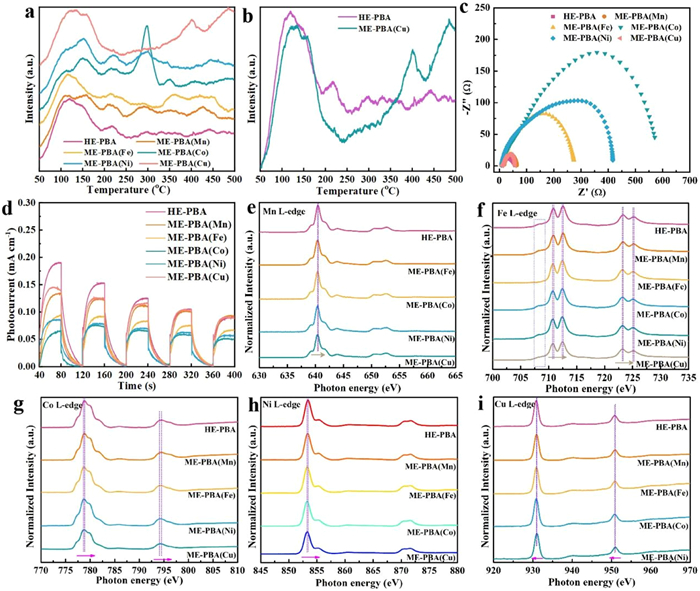
Charge separation efficiency was assessed using electrochemical impedance spectroscopy (EIS), an analysis of time-resolved photocurrent curves, and photoluminescence (PL) emission spectroscopy. The Nyquist plots (Fig. 4c and Fig. S14 in Supporting information) show a clear trend in the high-frequency semicircle diameters, increasing in the order: HE-PBA < ME-PBA(Cu) < ME-PBA(Mn) < ME-PBA(Fe) < ME-PBA(Ni) < ME-PBA(Co). This trend demonstrates that incorporating these five transition metals into PB enhances electrical conductivity by reducing charge transfer resistance. The migration of charge carriers in HE-PBA exhibits the lowest electron transport resistance, thus demonstrating a desirable electron transport capability. Furthermore, the HE-PBA delivered the optimal photocurrent response (Fig. 4d), indicating more efficient charge transport. Moreover, ME-PBA(Ni) and ME-PBA(Co) exhibited strong steady-state photoluminescence (PL) emission at approximately 440 nm, indicating inefficient charge transfer and rapid recombination of photogenerated charges with holes. In contrast, a marked fluorescence quenching was observed for HE-PBA, confirming a lower recombination of photo-generated electrons and holes (Fig. S15 in Supporting information), which suggests a longer carrier lifetime [40]. Based on these results, the HE-PBA system shows significant potential for promoting CO2RR.
Charge transfer was verified by measuring the metal L-edge using X-ray absorption near-edge structure (XANES) spectroscopy. As a result of 2p3/2 and 2p1/2 core electron spin-orbit interactions, the metal L-edge XANES spectra were split into L3 and L2 lines, reflecting the electronic dipole transition from the 2p core level to the 3d unoccupied state [42]. The Mn L-edge is consistent with a Mn2+ valence state, similar to the reported MnO XANES spectrum (Fig. 4e) [43]. In the case of the Fe L-edge spectra, the signals at 710–713 eV were attributed to Fe2+, whereas the appearance of a small shoulder at 708 eV implied the presence of the Fe3+ in addition to ME-PBA(Fe), confirming the conversion of Fe2+ to Fe3+ via electron loss (Fig. 4f) [44,45]. Furthermore, the appearance of four distinct peaks in the Co-L3 edge (777–782 eV, Fig. 4g) proved the coexistence of Co2+/Co3+ valence states [46]. Also, the main peak associated with Ni L3-edge at 853 eV was attributed to Ni2+, where a small peak at 855 eV indicated a higher oxidation state (Ni3+), as displayed in Fig. 4h [47-49]. The Cu XANES analysis (Fig. 4i) generated signals at 930.8 eV and 951.2 eV that were associated with Cu2+ species, whereas the peak at 939.7 eV represented a lower oxidation state, such as Cu+ [50]. The analysis suggests that Co, Ni, and Cu undergo valence changes as a result of electron transfer, where Co and Ni lose electrons and Cu gains electrons. Of interest, the Mn2+, Fe2+, Co2+, and Ni2+-related peaks in HE-PBA were slightly shifted to higher energies relative to ME-PBAs, conforming electron loss due to metal component interactions [44,45]. Moreover, the peaks associated with the HE-PBA Cu-L-edge exhibited a shift to a lower energy, consistent with Cu acting as an electron acceptor [29,30]. These results are in agreement with the XPS analysis.
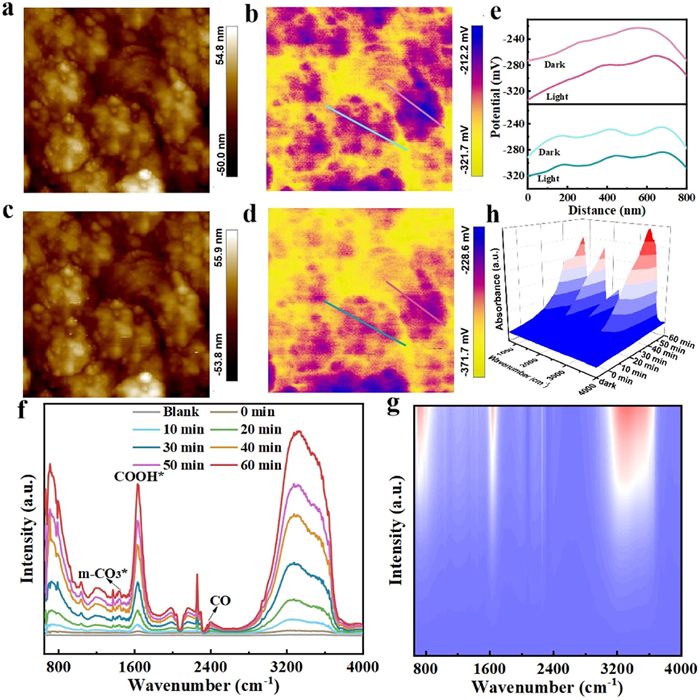
We investigated catalyst surface electronic properties to determine the direction of electron flow by Kelvin Probe Force Microscopy (KPFM) under dark and illuminated conditions. As shown in Figs. 5a-e and Figs. S16-S20 (Supporting information), all the synthesized catalysts exhibited a negative surface potential in the dark, indicating the presence of surface electron carriers. Following illumination, the ME-PBA(Mn), ME-PBA(Fe), ME-PBA(Ni), and ME-PBA(Cu) samples showed increased surface potentials, suggesting a greater tendency for internal electron flow with a resultant decrease in surface electron density. The HE-PBA and ME-PBA(Co) systems exhibited a common trend in terms of increased negative surface potentials, indicating an accumulation of electrons on the surface of the catalyst, forming an "electron-rich surface" [41]. More importantly, the HE-PBA catalyst showed the greatest change in surface potential (from −270 mV to −340 mV), due to a lower recombination of photo-generated electrons and holes, which should be in favor of promoting the photo-reduction of CO2.
From the diffuse reflectance UV–vis spectra in Fig. S21a (Supporting information), all samples exhibit similar absorption intensities in the UV region. However, in the visible and infrared regions, ME-PBA(Fe) shows significantly lower absorption strength compared to the other samples. This observation suggests that metallic iron is the primary component responsible for light absorption, which may contribute to enhancing photogenerated charge carriers to some extent [51]. The band gap energy (Eg) of the samples was determined via diffuse reflectance UV–vis spectroscopy (Fig. S20a). Applying the Kubelka-Munk function, the estimated Eg values for HE-PBA, ME-PBA(Mn), ME-PBA(Fe), ME-PBA(Co), ME-PBA(Ni), and ME-PBA(Cu) were 0.90, 0.88, 1.73, 0.86, 0.86, and 0.86 eV, respectively (Fig. S21b in Supporting information). The highest energy occupied molecular orbital (HOMO) positions derived from the XPS valence band spectra were 0.24, 0.28, 0.29, 0.26, 0.24, and 0.28 eV (Fig. S21c in Supporting information). Meanwhile, the calculated lowest energy unoccupied molecular orbital (LUMO) values were −0.66, −0.60, −1.44, −0.60, −0.62, and −0.58 eV, respectively [52]. The schematic representation presented in Fig. S21d (Supporting information) illustrated the energy band structure. It was evident that the LUMO values of all the catalysts were more negative than the reduction potential for CO2 to CO (−0.53 eV), revealing that the catalysts can effectively promote the photocatalytic reduction of CO2.
The surface CO2RR on HE-PBA was probed by in situ diffuse reflectance infrared Fourier transform spectroscopy (in situ DRIFTS) to dynamically monitor the adsorbed surface species and CO2-derived intermediates. A baseline correction was applied to all spectra in order to exclude environmental factors. The spectra for the photocatalyst prior to irradiation were set as the background. As shown in Figs. 5f-h, contacting HE-PBA with a mixture of CO2 and H2O vapors resulted in distinct signals peaks at 1415 and 1533 cm-1 that can be attributed to monodentate carbonate (m-CO3*) and bidentate carbonate (b-CO3*) species, respectively [53]. Meanwhile, surface bicarbonate (HCO3*) bending modes give rise to peaks at 1440 cm-1, originating from the adsorption of CO2 and -OH groups [54]. It should be noted that the intensity of the HCO3* peak increased with irradiation time, suggesting a role as intermediates for the subsequent production of C1 fuel. Most importantly, the COOH* species, with characteristic peak at 1643 cm-1, represents the most critical intermediate in the photocatalytic conversion of CO2 to CO [55]. Furthermore, the weak absorption intensities at 2416 cm-1 are associated with CO*, which is directly related to the generation of the final CO product [56,57]. Based on an analysis of the in situ DRIFTS, we proposed that the CO2 photo-reduction pathway over HE-PBA proceeded as: CO2 → HCO3* → HCOO* → CO* →CO.
In conclusion, in response to global concerns regarding greenhouse gases such as CO2, which contribute to the greenhouse effect and energy crisis, photocatalysis has emerged as a viable technology to achieve "carbon neutrality". Catalyst design is essential to ensure process efficiency. This study had demonstrated that high-entropy Kx(MnFeCoNiCu)[Fe(CN)6] (HE-PBA) was effective in mediating CO2 adsorption, activation and reduction. The application of HE-PBA ensured a high concentration of active sites and an effective separation of photo-generated charge carriers. Meanwhile, the photo-generated electrons migrated and accumulated on the HE-PBA surface, forming a local electron-rich environment that served to enhance CO2 reduction. Under light irradiation with CO2 as the sole carbon source, HE-PBA achieved a CO2-to-CO conversion yield of 1220.5 µmol g-1 h-1, outperforming medium-entropy ME-PBA(Mn), ME-PBA(Fe), ME-PBA(Co), ME-PBA(Ni), and ME-PBA(Cu) catalysts by a factor of two-fold, three-fold, four-fold, four-fold, and two-fold, respectively.
Given the range of non-noble metal elements in high-entropy materials, there is considerable scope for enhancing catalyst performance through rational design and an informed choice of metal components to ensure effective coupling to adjust the electronic character and enhance charge transfer, optimizing photocatalytic performance. The findings of this study can inform the design of advanced high-entropy photocatalysts for environmental remediation applications. Several issues must be addressed, notably the requirement for a sacrificial reducing agent, which represents a critical shortcoming. Coupling CO2RR with a water oxidation reaction system can address this issue and represents a topic for future research.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Xiaohong Li: Writing – original draft, Data curation, Conceptualization. Limin Jin: Validation, Formal analysis. Haihui Yu: Software, Resources. Yuteng Zhang: Validation, Methodology. Haifeng Zhang: Writing – review & editing, Project administration, Funding acquisition. Yanbiao Liu: Writing – review & editing, Supervision.
This work was financially supported by the National Natural Science Foundation of China (Nos. 52070035, 22401039), Jilin Province Scientific and the Technological Planning Project of China (No. 20200403001SF), Science and Technology Project of Jilin Education Department (No. JJKH20250853KJ), and Start-up Fund for Doctoral Research of Northeast Electric Power University (No. BSJXM-2024112).
Supplementary material associated with this article can be found, in the online version, at doi:
S.M. Wang, D.X. Yan, W.H. Zhang, et al., Chin. Chem. Lett. 36 (2025) 110611. doi: 10.1016/j.cclet.2024.110611
E.V. Roijen, S.A. Miller, S.J. Davis, Science 387 (2025) 176–182. doi: 10.1126/science.adq8594
W.D. Meng, Y.B. Zhou, Y. Zhou, Chin. Chem. Lett. 36 (2025) 109961. doi: 10.1016/j.cclet.2024.109961
B. Zeng, F.W. Huang, Y.X. Wang, Chin. J. Catal. 58 (2024) 226–236. doi: 10.1016/S1872-2067(23)64601-7
N. Sakamoto, K. Sekizawa, S. Shirai, et al., Nat. Catal. 7 (2024) 574–584. doi: 10.1038/s41929-024-01147-y
J.C. Wu, F. Huang, Q.Y. Hu, et al., J. Am. Chem. Soc. 146 (2024) 26478–26484. doi: 10.1021/jacs.4c09841
Q. Zhang, S.Q. Gao, Y.Y. Guo, et al., Nat. Commun. 14 (2023) 1147. doi: 10.1038/s41467-023-36779-4
B. Zeng, Y.X. Wang, F.W. Huang, et al., Catal. Sci. Technol. 14 (2024) 2838–2847. doi: 10.1039/D4CY00312H
Z.G. Guo, G. Chen, C. Cometto, et al., Nat. Catal. 2 (2019) 801–808. doi: 10.1038/s41929-019-0331-6
W. Jiang, T. Wang, H. Chen, et al., Nano Energy 79 (2021) 105464. doi: 10.1016/j.nanoen.2020.105464
X.F. Chen, C.D. Peng, W.Y. Dan, et al., Nat. Commun. 13 (2022) 4592. doi: 10.1038/s41467-022-32367-0
H.G. Jin, P.C. Zhao, Y.Y. Qian, et al., Chem. Soc. Rev. 53 (2024) 9378. doi: 10.1039/D4CS00095A
W.L. Sheng, F.W. Huang, X.J. Lang, Mater. Today Chem. 30 (2023) 101505. doi: 10.1016/j.mtchem.2023.101505
A. Gouda, K. Hannouche, A. Mohan, et al., Nat. Commun. 16 (2025) 695. doi: 10.1038/s41467-025-55891-1
Z. Jiang, X.H. Xu, Y.H. Ma, et al., Nature 586 (2020) 549–554. doi: 10.1038/s41586-020-2738-2
K. Song, S. Liang, X. Zhong, et al., Appl. Catal. B 309 (2022) 121232. doi: 10.1016/j.apcatb.2022.121232
Y.R. Ji, Y.F. Guo, X. Liu, et al., Chem. Eng. J. 471 (2023) 144743. doi: 10.1016/j.cej.2023.144743
C. Wei, X.Y. Fu, L.L. Zhang, et al., Chem. Eng. J. 421 (2021) 127760. doi: 10.1016/j.cej.2020.127760
J.H. Jeoung, H. Dobbek, Science 318 (2007) 1461–1464. doi: 10.1126/science.1148481
A.M. Appel, J.E. Bercaw, A.B. Bocarsly, et al., Chem. Rev. 113 (2013) 6621–6658. doi: 10.1021/cr300463y
H.Y. Yang, G.W. Liu, L.X. Zheng, et al., Appl. Catal. B 359 (2024) 124491. doi: 10.1016/j.apcatb.2024.124491
X.S. Gao, B. Guo, C.M. Guo, et al., ACS Appl. Mater. Interfaces 12 (2020) 24059–24065. doi: 10.1021/acsami.0c05631
H.H. Fei, M.D. Sampson, Y. Lee, et al., Inorg. Chem. 54 (2015) 6821–6828. doi: 10.1021/acs.inorgchem.5b00752
Y.J. Ma, Y. Ma, S.L. Dreyer, et al., Adv. Mater. 33 (2021) 2101342. doi: 10.1002/adma.202101342
H.W. Huang, J.W. Zhao, H.L. Guo, et al., Adv. Mater. 36 (2024) 2313209. doi: 10.1002/adma.202313209
X.Y. Meng, J.Y. Yang, C.C. Zhang, et al., ACS Catal. 12 (2022) 89–100. doi: 10.1021/acscatal.1c04415
Y.J. Ma, Y. Ma, S.L. Dreyer, et al., Adv. Mater. 33 (2021) 2101342. doi: 10.1002/adma.202101342
H.W. Huang, J.W. Zhao, H.L. Guo, et al., Adv. Mater. 36 (2024) 2313209. doi: 10.1002/adma.202313209
L.N. Tang, Y.L. Yang, H.Q. Guo, et al., Adv. Funct. Mater. 32 (2022) 2112157. doi: 10.1002/adfm.202112157
M.L. Huang, J. Lu, J.X. Ji, et al., Chem. Eng. J. 485 (2024) 149633. doi: 10.1016/j.cej.2024.149633
S. Zaman, Y.Q. Su, C.L. Dong, et al., Angew. Chem. Int. Ed. 61 (2022) e202115835. doi: 10.1002/anie.202115835
J. Lu, X. Xu, H.W. Zhang, et al., Sensor. Actuat. B: Chem. 422 (2025) 136605. doi: 10.1016/j.snb.2024.136605
X.F. Bie, K. Kubota, T. Hosaka, et al., J. Power Sources 378 (2018) 322. doi: 10.1016/j.jpowsour.2017.12.052
M.Y. Liao, Y.N. Cai, L. Chen, et al., Sep. Purif. Technol. 337 (2024) 126410. doi: 10.1016/j.seppur.2024.126410
R.X. Huang, M. Xie, J.T. Zhang, et al., Nano Energy 39 (2017) 273. doi: 10.1016/j.nanoen.2017.07.005
Q.S. Wang, A. Sarkar, D. Wang, et al., Energy Environ. Sci. 12 (2019) 2433. doi: 10.1039/C9EE00368A
Y.B. Zhao, B.L. Liang, X.J. Wei, et al., J. Mater. Chem. A 7 (2019) 10464. doi: 10.1039/C8TA12433G
X.K. Zhang, M.T. Xia, T.T. Liu, et al., Chem. Eng. J. 421 (2021) 127767. doi: 10.1016/j.cej.2020.127767
A. Li, Q. Cao, G.Y. Zhou, et al., Angew. Chem. Int. Ed. 58 (2019) 14549–14555. doi: 10.1002/anie.201908058
C. Chen, M.G. Wu, Y.F. Xu, et al., J. Am. Chem. Soc. 146 (2024) 9163–9171. doi: 10.1021/jacs.3c14590
X.H. Li, L. Yang, Q.L. Liu, et al., Adv. Mater. 35 (2023) 2304532. doi: 10.1002/adma.202304532
Y.L. Ma, Y. Zhou, C.Y. Du, et al., Chem. Mater. 29 (2017) 2141–2149. doi: 10.1021/acs.chemmater.6b04784
B. Gilbert, B.H. Frazer, A. Belz, et al., J. Phys. Chem. A 107 (2003) 2839–2847. doi: 10.1021/jp021493s
L. Wu, C.J. Li, M.F. Chen, et al., ACS Appl. Mater. Interfaces 9 (2017) 44931–44937. doi: 10.1021/acsami.7b15364
J.P. Wu, J. Song, K.H. Dai, et al., J. Am. Chem. Soc. 139 (2017) 18358–18364. doi: 10.1021/jacs.7b10460
Y. Zhang, Y.X. Hu, Z.L. Wang, et al., Adv. Funct. Mater. 30 (2020) 2004172. doi: 10.1002/adfm.202004172
K. Shimoda, K. Yazawa, T. Matsunaga, et al., Sci. Rep. 10 (2020) 10048. doi: 10.1038/s41598-020-66411-0
F. Lin, D. Nordlund, I.M. Markus, et al., Energy Environ. Sci. 7 (2014) 3077–3085. doi: 10.1039/C4EE01400F
H.W. Chang, C.H. Lee, Y.X. Hong, et al., Energies 16 (2023) 5467. doi: 10.3390/en16145467
Ł. Kuterasiński, J. Podobiński, E. Madej, et al., Molecules 25 (2020) 4765. doi: 10.3390/molecules25204765
S. Barman, A. Singh, F.A. Rahimi, et al., J. Am. Chem. Soc. 143 (2021) 16284. doi: 10.1021/jacs.1c07916
X.H. Li, H. Li, S.L. Jiang, et al., ACS Catal. 13 (2023) 7189–7198. doi: 10.1021/acscatal.3c00944
L.Y. Wu, Y.P. Li, B.H. Zhou, et al., Carbon 207 (2023) 36–48. doi: 10.1016/j.carbon.2023.03.003
H.B. Yin, J.H. Li, Appl. Catal. B 320 (2023) 121927. doi: 10.1016/j.apcatb.2022.121927
F. Raziq, C.Y. Feng, M. Hu, et al., J. Am. Chem. Soc. 146 (2024) 21008–21016. doi: 10.1021/jacs.4c05873
X.D. Li, L. Li, G.B. Chen, et al., Nat. Commun. 14 (2023) 4034. doi: 10.1038/s41467-023-39666-0
Y. Wang, J.X. Wei, H.L. Tang, et al., Nat. Commun. 15 (2024) 8818. doi: 10.1038/s41467-024-53066-y

Figure 1 Schematic diagram of reaction process. Schematic representation of CO2 activation and conversion to CO by HE-PBA.

Figure 2 (a) Schematic representation of HE-PBA synthesis. (b) XRD patterns for HE-PBA, ME-PBA(Mn), ME-PBA(Fe), ME-PBA(Co), ME-PBA(Ni), and ME-PBA(Cu). (c-e) HR-TEM, spherical aberration corrected HAADF-STEM and TEM characterization of HE-PBA. (f-j) Mn 2p, Fe 2p, Co 2p, Ni 2p, and Cu 2p XPS spectra.

Figure 3 (a) CO and H2 products yield and (b) CO product yield over HE-PBA, ME-PBA(Mn), ME-PBA(Fe), ME-PBA(Co), ME-PBA(Ni), and ME-PBA(Cu). (c) CO and H2 selectivity. (d) Control experiments for HE-PBA. (e) Synchrotron-radiation vacuum ultraviolet photoionization mass spectrometry (SVUV-PIMS) pattern for the product generated in the photocatalytic reduction of 13CO2 over HE-PBA. (f) Cycling experiments over HE-PBA.

Figure 4 (a) CO2-TPD profiles for the synthesized catalysts. (b) CO2-TPD profiles for HE-PBA and ME-PBA(Cu) as a contrast. (c) EIS Nyquist plots. (d) Time-resolved photocurrent curves. (e-i) XANES spectra of Mn L-edge, Fe L-edge, Co L-edge, Ni L-edge, and Cu L-edge.

Figure 5 AFM and KPFM potential images of HE-PBA under dark (a, b) and light (c, d) conditions. (e) The corresponding potential section of the line under dark and light conditions. (f) In situ DRIFTS spectra recorded during photocatalytic CO2 reduction over HE-PBA in the presence of water vapor. The corresponding 2D contour color fill plots (g) and 3D RL plots (h) for HE-PBA.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
 下载:
下载: