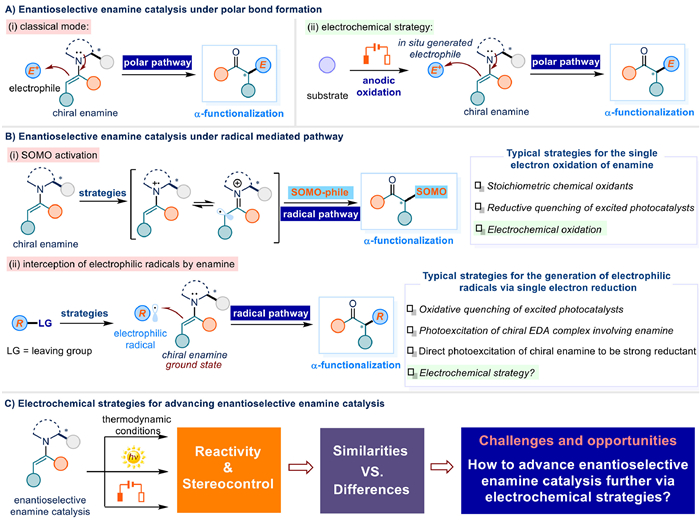
Figure 1.
Overview of enantioselective enamine catalysis.

The enantioselective synthesis of functionalized carbonyl compounds continues to represent a fundamental research priority across chemical sciences, with particular significance in synthetic methodology development and pharmaceutical manufacturing [1–4]. Within this context, enamine catalysis has emerged as a pivotal strategy, deriving its value from a synergistic combination of stereochemical precision, operational efficiency, and sustainable practice. The classical paradigm of enantioselective enamine catalysis involves stereodefined bond formation (C–C/C-X) through nucleophilic engagement of chiral enamine intermediates with electrophilic partners, effectively establishing α-stereocenters in carbonyl frameworks [4–16]. This polar chemical bond formation mechanism (Fig. 1A-i) operates through directed electron pair transfer from the electron-rich enamine species to the electrophilic counterpart, enabling predictable stereochemical outcomes.
In continuous developments, the strategic implementation of radical mediated chemical bond construction via radical species has expanded the synthetic utility of enamine systems. Current mechanistic frameworks can be predominantly classified into two distinct categories based on enamine intermediate forms: (ⅰ) Stereocontrolled coupling of SOMO-philes with chiral enamine radical cations generated through single-electron oxidation (Fig. 1B-i) [17–19]. (ⅱ) Stereoselective interception of electrophilic radicals by ground state enamine species [19,20], triggered by single electron reduction from excited photocatalysts [19,21–25], photoexcitation of chiral EDA complex including chiral enamine [26], direct photoexcitation of chiral enamine to be a strong single electron reductant (Fig. 1B-ⅱ) [27–29].
Recent years, synthetic electrochemistry has undergone significant growth. By harnessing electrical energy as a traceless redox equivalent, this approach fundamentally circumvents the reliance on stoichiometric chemical oxidants or reductants for traditional thermodynamic reactions, thereby aligning with green chemistry principles. This paradigm shift not only enhances synthetic efficiency but also expands the chemical space accessible for precision molecular editing and functional group manipulation [30–44]. Within enantioselective enamine catalysis, its combination with electrochemistry also brings many innovative transformations in line with the goal of acquisition optical α-stereocentered carbonyl compounds in a more efficient, economic, and greener manner.
So far, electrochemical enamine catalysis has seen its success in above mentioned polar and radical mediated transformations. For instance, electrochemical oxidation converts substrates to active electrophiles via multi-electron transfer; these species then react with chiral enamines through polar pathways to deliver products (Fig. 1A-ⅱ). Meanwhile, the electrochemically facilitated SOMO activation also emerged, demonstrating the capability of electrochemical methods to extend beyond conventional approaches reliant on stoichiometric chemical oxidants or photoexcited catalysts (Fig. 1B-i).
Notably, both photonic and electrical energy inputs are increasingly recognized as clean energy sources, positioned as sustainable drivers for next-generation chemical manufacturing. Significantly, the strategic integration of these energy modalities with enamine catalysis has established a continuously evolving platform for constructing chiral carbonyl compounds bearing α-stereocenters with enhanced precision and environmental compatibility. While previous reviews have systematically summarized light-mediated asymmetric enamine catalysis [19,20], electrochemical enantioselective enamine catalysis lacks integral reviews to outline its full development at this moment [45–53].
This review specifically focused on advancements in electrochemical activations concerning three principal targets (Fig. 1C): (a) Highlighting redox-stereochemical synergy within electrochemical enantioselective enamine catalysis, and pointing out crucial issues such as electrode materials, electrolyte selection, and additives in the optimization, which would be helpful for researchers to understand mechanistic paradigms and experimental challenges associated with electrochemical enantioselective enamine catalysis beyond thermodynamic and photochemical conditions; (b) Recognizing the true value of electrochemistry lies in enabling enhanced reactivities rather than mimicking traditional oxidants; (c) Inspiring potential development avenues by addressing the challenges in this area, including but not limited to the integration of electrophilic radicals with chiral enamine via cathodic reduction (Fig. 1B-ⅱ).
To differentiate between electrochemical enantioselective enamine catalysis pathways, polar bond formation versus radical-mediated mechanisms, we provide a schematic outline prior to discussing individual reactions. The operative mechanism hinges on the oxidation state of the enamine during the key bond-forming step. When the chiral enamine has a low oxidation potential tending to be oxidized, single-electron oxidation generates a radical cation intermediate, which exists in equilibrium with the α-imino radical. Notably, the single electron oxidation occurred either at the anode or initiated by the redox mediator/shuttle homogeneously. In this scenario, the α-imino radical intermediate reacts with SOMO-philes to afford the product via a radical-mediated pathway. Key mechanistic identification methods include: Cyclic voltammetry (CV) revealing a low oxidation potential for the enamine, signifying its susceptibility to oxidation; Radical scavengers (e.g., BHT, O2) suppressing or halting product formation; Radical clock experiments using β-cyclopropyl-substituted carbonyls, evidenced by cyclopropane ring-opening; Exclusion studies directly reacting the enamine with potential electrophiles, and no product formation under this condition makes a polar pathway less likely (Fig. 2a).
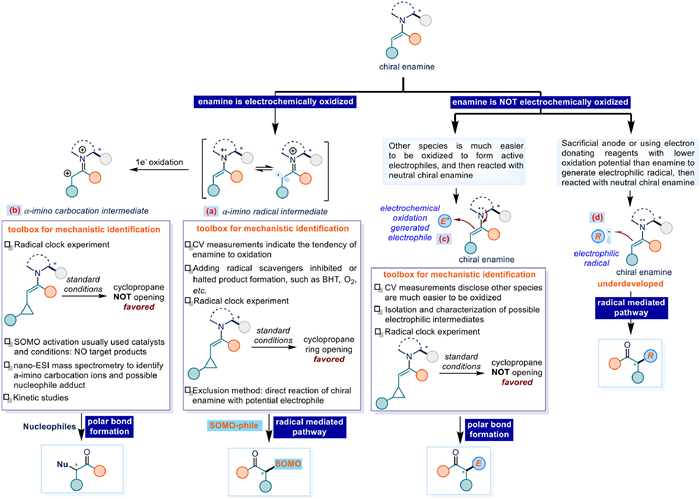
Following a subsequent single-electron oxidation, the α-imino radical intermediate converts to an α-imino cation intermediate. This electrophilic intermediate then couples with diverse nucleophiles via an SN1 pathway, representing a polar bond-formation process involving the enamine species. Key mechanistic evidence includes: Radical clock experiments using β-cyclopropyl-substituted carbonyls, where the absence of cyclopropane ring-opening would support this mechanism; Catalysts or conditions for SOMO-activation failed to afford the target products in this scenario would also provide positive mechanistic support; Mass spectrometric detection of α-imino cation intermediates or their nucleophile adducts; Kinetic studies could bring additional mechanistic insights (Fig. 2b).
When other compounds in the electrochemical cell with significantly lower oxidation potentials are present, the chiral enamine remains in its neutral form. Under these conditions, two distinct reaction modes emerge. On one side, reagents undergo successive/multiple electron oxidation to generate active electrophilic species. These are then captured by the neutral enamine through polar bond formation, yielding the product. The aminocatalyst provides a well-defined chiral environment, ensuring highly stereoselective coupling. Within this reaction mode, several methods would be helpful for the mechanistic investigation: CV tests could recognize the species more susceptible to oxidation than enamine; Isolation and characterization of any potential electrophilic intermediates would be direct evidence; Radical clock experiments with β-cyclopropyl carbonyl compounds show absence of ring-opening, supporting non-radical mechanism (Fig. 2c).
On the other side, electrophilic radicals might be generated from corresponding precursors via electrochemical reduction promoted by sacrificial anode or anodic oxidation of electron donating reagents featuring lower oxidation potential than enamine. Next, this electrophilic radical could be trapped by the native enamine via a radical mediated pathway to yield α-functionalized products. However, this mechanistic proposal under electrochemical conditions remains underdeveloped, with stereoselectivity challenges requiring further validation (Fig. 2d). Notably, the following reactions all use undivided cells, and this will not be repeated in each reaction description.
In contemporary electrochemical enantioselective catalysis mediated by enamine catalysis, the predominant paradigms continue to involve coupling the native enamine species with electrogenerated electrophiles through anodic oxidation. However, this approach presents critical challenges in maintaining the oxidative stability of reaction components, particularly the aminocatalysts and chiral enamine intermediates [54,55], which requires careful evaluation during electrochemical system design. Notably, while the polar bond formation and stereocontrol mechanism have been considered quite reliable in conventional thermodynamic processes, its efficacy in electro-chemical environments demands re-examination. The following successful case studies demonstrate the enduring potential of the polar pathway in modern electrochemical transformations.
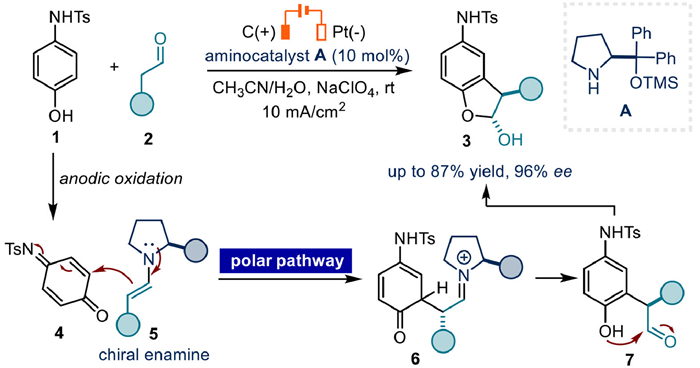
In 2010, Jørgensen's group reported an electrochemical enantioselective synthesis of meta-alkylated anilines via organocatalytic α-arylation of enamine [56]. Mechanistically, it is the N-(4-hydroxyphenyl)-4-methylbenzenesulfonamide 1 to undergo anodic oxidation to yield electrophilic intermediate 4, then reacted with chiral enamine 5 to afford intermediate 6. Subsequent isomerization and hydrolysis would form 7. Finally, hemi-acetal product 3 could be generated and isolated through intramolecular cyclization of 7. Notably, the key bond formation step proceeds through a polar process (Fig. 3).
After that, Luo's group reported an electrochemical oxidative cross-dehydrogenative coupling reaction of tetrahydroisoquinoline 8 and simple ketone 9 to afford C1-alkylated tetrahydroisoquinoline derivatives 10 in high enantioselectivity in 2017 [57]. Cyclic voltammetry measurements and other control experiments indicated that tetrahydroisoquinoline 8 was transferred to active iminium ion intermediate 11 via anodic oxidation, ensuring a following stereoselective capture by chiral enamine 12 to give the final product 10. In this case, the crucial bond construction step is also a polar pathway (Fig. 4a).
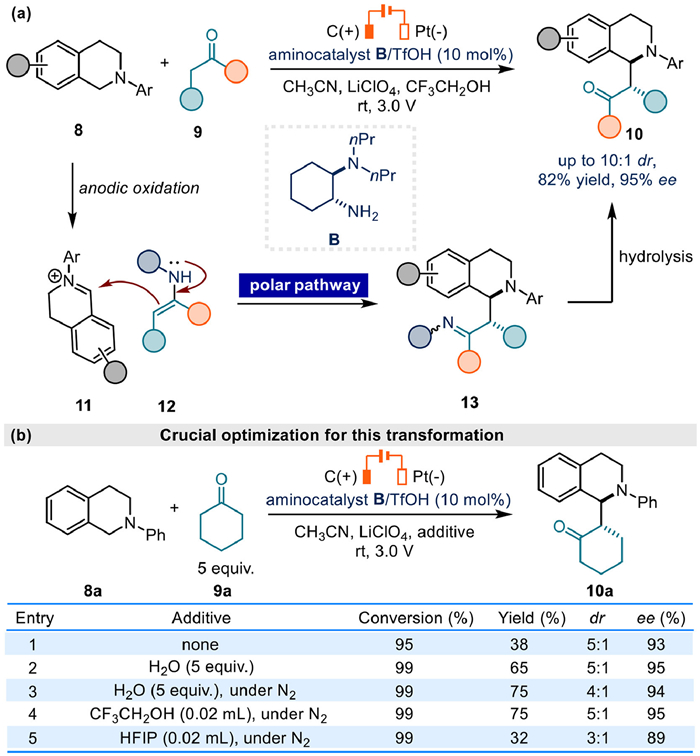
In the initial optimization, the authors observed high conversion but low yield (Fig. 4b, entry 1). Subsequently, various protonic additives were evaluated to improve efficiency. These additives potentially serve two functions: stabilizing the in situ generated iminium ion intermediate through hemiaminal formation [58,59], and enhancing conductivity via the cathodic hydrogen evolution. Upon adding 5 equiv. of water, the yield increased to 65% (Fig. 4b, entry 2). Further optimization under a N2 atmosphere afforded a higher yield of 75%, albeit with slightly reduced dr and ee values (Fig. 4b, entry 3). Ultimately, CF3CH2OH proved most effective, while 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) was less effective (Fig. 4b, entries 4 and 5).
As a comparison, this transformation was reported by Wang et al. using stoichiometric DDQ as the chemical oxidant in combination with chiral amino acid catalyst C, proposed to proceed via an enamine-involved chiral ion pair pathway (Fig. 5a) [60]. In 2017, Wu and Luo developed a photoredox approach employing synergistic triple catalysis (enamine/Ru/Co), where substoichiometric m-nitrobenzoic acid was required for cobalt cycle completion (Fig. 5b) [61]. Subsequently, Rueping et al. reported an alternative photoredox system using catalytic aminocatalyst D to deliver moderate yields with good enantioselectivity. In this reaction, an oxygen atmosphere was required (Fig. 5c) [62].
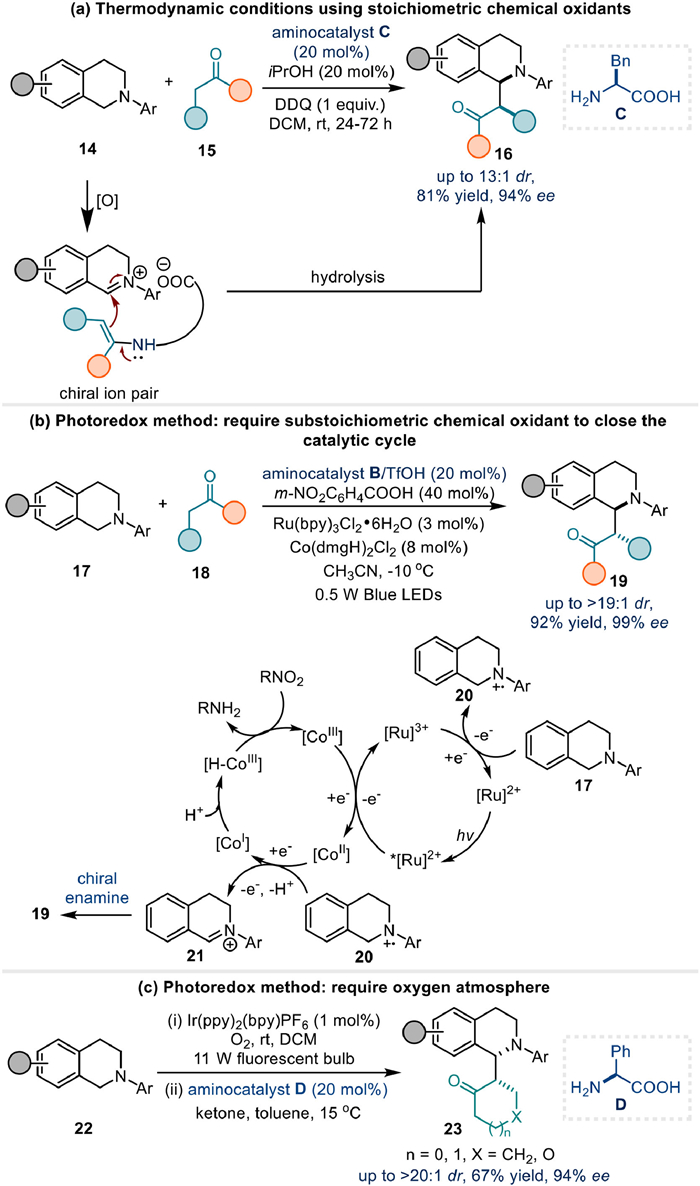
C2-fully substituted indolin-3-ones are important scaffolds in many natural products. He's group developed an electrochemical enantioselective construction of indolin-3-ones 26 bearing 2-quaternary stereocenters in high diastereoselectivity and enantioselectivity via organocatalytic enamine catalysis from 2-aryl indole 24 and simple ketone 25 (Fig. 6a) [63]. Mechanistic experiment revealed that 3H-indol-3-one 33 was a possible electrophilic intermediate to react with chiral enamine 34, affording intermediate 35 through polar process. Final hydrolysis of 35 would provide the expected product 26. In addition, control experiment indicated that the TEMPO additive was helpful to improve the yield of 26a from to 50% to 67%. Therefore, a plausible mechanism was then proposed in combination with the cyclic voltammetry measurements. It is the TEMPO to undergo single electron oxidation at the anode to form TEMPO+ 27 firstly, and then indole substrate 24 was oxidized by TEMPO+ through SET (single electron transfer) to yield 28. After losing a proton, radical intermediate 29 and its resonance 30 were generated.
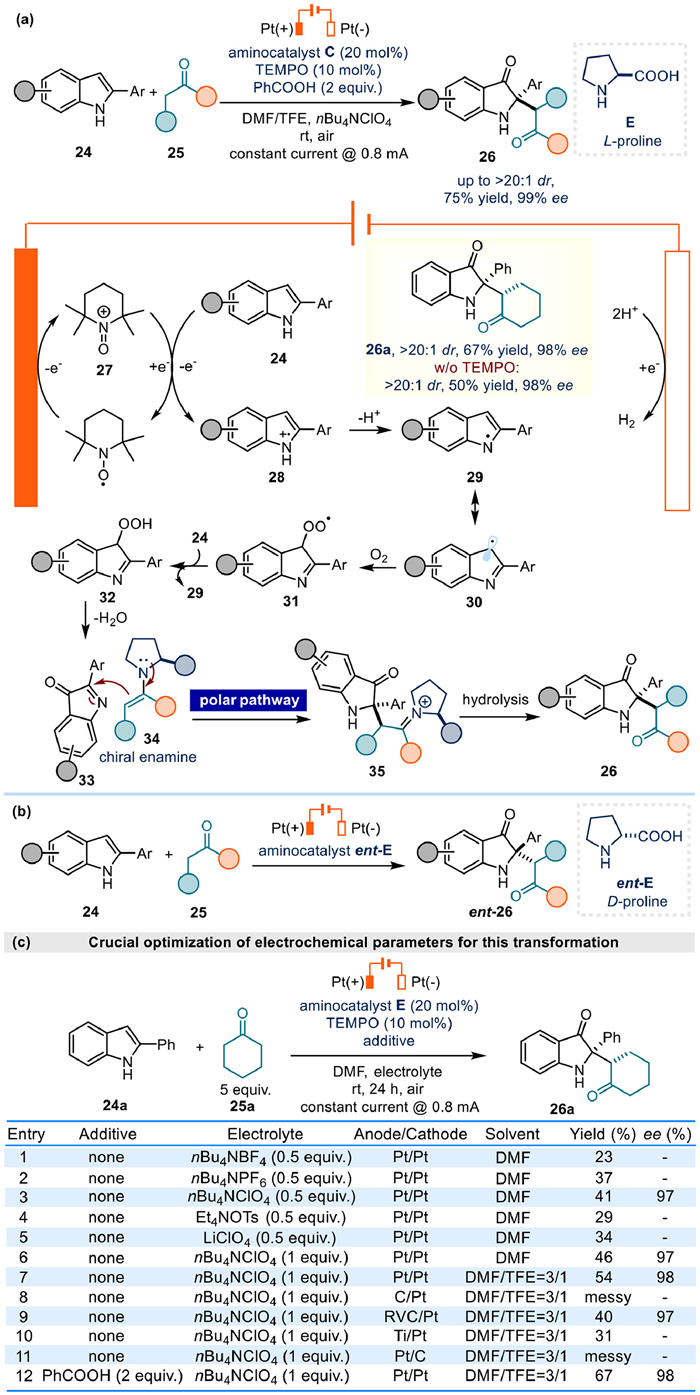
Meanwhile, O2 is proved to be quite important for this reaction since the yield of 26a would drop significantly in argon atmosphere. The authors inferred that the oxygen of product 26a might come from O2 in the air atmosphere. And this hypothesis was then confirmed by 18O2 labelling experiment. Thus, radical intermediate 30 should react with O2 next to afford radical intermediate 31, followed by the HAT (hydrogen atom transfer) from indole 24 to give intermediate 32, together with the generation of intermediate 29. Subsequent elimination of water from 32 would yield the key electrophile 33.
Notably, catalytic employment of D-proline (ent–E) enabled stereodivergent synthesis of the enantiomers of product 26 (Fig. 6b). This achievement not only establishes configurational diversity in C2-fully substituted indolin-3-one systems but also critically validates the robustness of the stereocontrol paradigm under electrochemical conditions.
When optimizing conditions, electrochemical parameters, specifically the electrolyte and electrodes, significantly influenced the outcome (Fig. 6c). Among those screened, nBu4NClO4 proved optimal as the electrolyte (Fig. 6c, entries 1–5), while Pt electrodes for both anode and cathode delivered superior results (Fig. 6c, entries 7–11). Employing a mixed DMF/TFE solvent system increased the yield (Fig. 6c, entries 6 and 7). Finally, adding PhCOOH further improved the yield to 67% (Fig. 6c, entry 12), probably by promoting enamine formation and providing protons for hydrogen evolution at the cathode.
Prior to the development of electrochemical methods, thermodynamic methods and photoredox catalysis have demonstrated their ability to promote this type of transformation. Notably, stoichiometric TEMPO+OTf- was employed to form the crucial intermediate 40 [64], which could also generate the hemiaminal intermediate 41 in the presence of water (Fig. 7a). The photochemical reaction, meanwhile, was conducted under an oxygen atmosphere with an elevated loading of aminocatalyst E [65]. The same electrophilic species 33 was formed either by single-electron reduction of oxygen gas to superoxide anion or via energy transfer (ET)-enabled [2 + 2] cycloaddition of singlet oxygen with indole reactants (Fig. 7b).
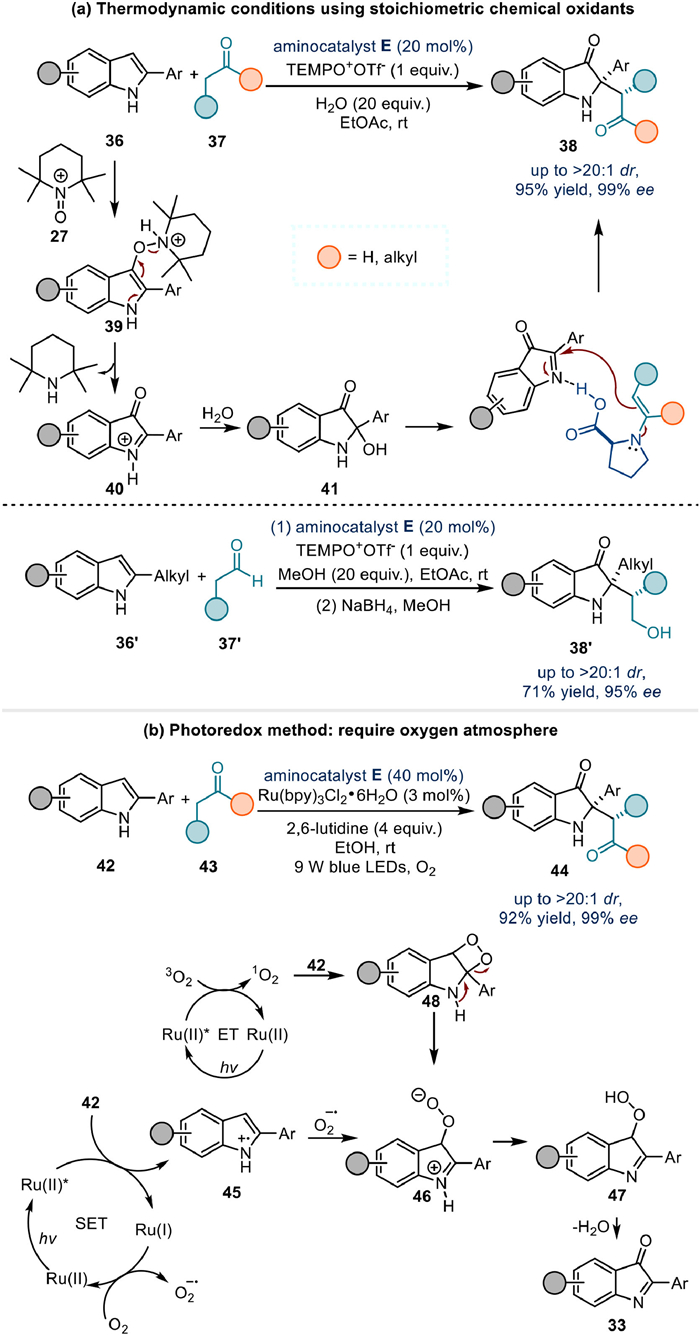
In 2020, Luo and colleagues reported a groundbreaking electrochemical strategy for the asymmetric α-arylation of cyclic β-ketocarbonyls using enamine catalysis (Fig. 8a) [66]. Central to this methodology was the in situ electrochemical generation of reactive benzyne 52, which proved critical for achieving high reaction efficiency. Conventional approaches employing 2-(trimethylsilyl)phenyl triflate 54 as a benzyne precursor under thermodynamic conditions could only yield < 10% product, underscoring the superiority of the electrochemical protocol. Mechanistically, anodic oxidation of 1-aminobenzotriazole 49 generated benzyne 52, which underwent stereoselective polar C–C bond formation with chiral enamine intermediate 53 to deliver product 51 with excellent enantioselectivity.
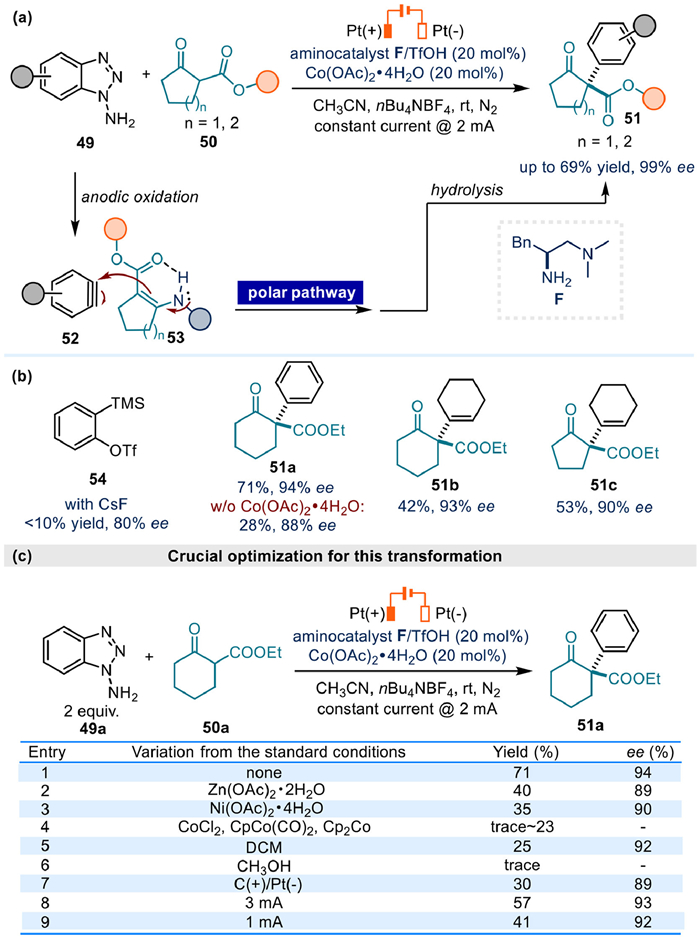
Cyclic voltammetry analysis rationalized the chemo-selectivity: 1-aminobenzotriazole 49a (Eox = 0.84 V) exhibited significantly lower oxidation potential compared to both the aminocatalyst (Eox = 1.54 V) and enamine intermediate (Eox = 1.15 V), ensuring preferential benzyne generation. The catalytic system further required Co(OAc)2•4H2O as a crucial additive, with control experiments demonstrating a dramatic decline in yield (28% vs. 71%) and slightly lower enantioselectivity (88% ee vs. 94% ee) in its absence (Fig. 8b). Benzyne quenching studies and DFT calculation suggested cobalt's role in stabilizing the transient aryne intermediate through triple-bond coordination.
Notably, apart from the capture of benzyne intermediate, α-vinylated products 51b and 51c could also be afforded starting from corresponding cyclic alkyne precursors in moderated yields and excellent ee values (Fig. 8b). This work establishes electrochemistry as a powerful tool for manipulating reactive intermediates in asymmetric catalysis.
Concerning the acquisition of the best conditions, metal additives (Fig. 8c, entries 2–4) and solvents (Fig. 8c, entries 5 and 6) both had an important effect. In addition, changing the anode material to graphite (Fig. 8c, entry 7), and varying currents (Fig. 8c, entries 8 and 9) all result in obviously diminished yields.
In 2021, Mei's team successfully realized the stereoselective Shono-type cross dehydrogenative coupling of acyclic amines and cyclic ketones by employing TEMPO as a redox mediator (Fig. 9) [54]. Notably, a suitable redox mediator was proved to be able to accelerate the electron transfer on the electrode surface, thus enables the electrochemical transformation at a lower oxidative potential, enhancing the overall efficiency and feasibility of the reaction process [39,67–69].
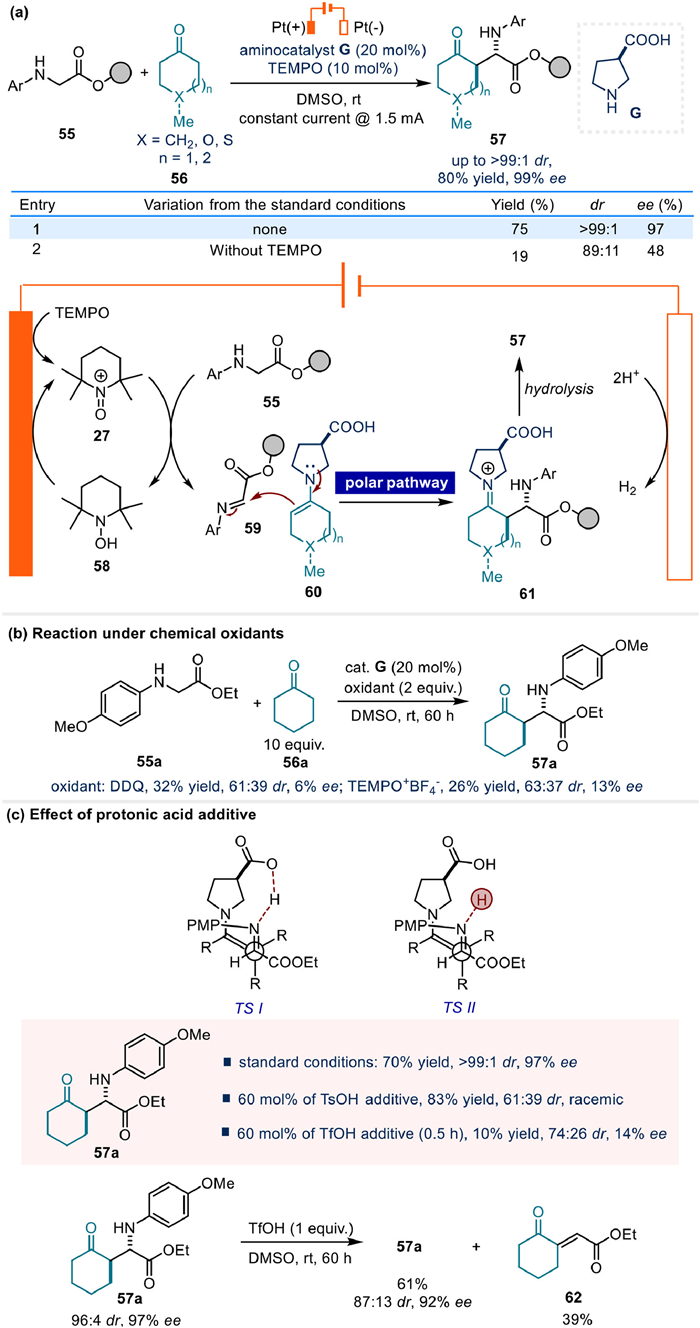
The crucial role of TEMPO in this reaction could be verified by the control experiment in the absence of it, which resulted in a significantly lower yield, as well as diastereo- and enantioselectivity (Fig. 9a). Meanwhile, cyclic voltammetry experiments also elucidated the mediator role of TEMPO. Based on these experimental findings, a plausible reaction mechanism was proposed. Initially, TEMPO undergoes single-electron oxidation at the anode to form the intermediate 27. Subsequently, 27 oxidizes glycine ester 55, yielding the electron-neutral imine intermediate 59. Following this, the nucleophilic addition of a chiral enamine to intermediate 59 generates intermediate 61. Ultimately, the hydrolysis step affords the target product 57 with high diastereo- and enantioselectivity. Moreover, it is noteworthy that the key C–C bond connection step in this transformation follows a polar pathway.
The significant advantages of the electrochemical strategy in this transformation are further highlighted by comparative studies. When chemical oxidants such as DDQ or TEMPO+BF4− were used instead of the electrochemical approach, the resulting products exhibited considerably lower diastereo- and enantioselectivity (Fig. 9b). Furthermore, the authors propose that a hydrogen bonding interaction between the acidic proton of chiral aminocatalyst G and the imine is crucial for achieving high enantioselectivity (TS-I, Fig. 9c). In contrast, alternative proton sources, such as protons generated from glycine ester 55 oxidation under thermodynamic conditions using chemical oxidants, could induce racemization due to competing interactions (TS-II, Fig. 9c). Further mechanistic investigations, including studies on the effects of proton additives and the stability of the products under acidic conditions, supported the above proposal and revealed that the key to the success of this protocol lies in the hydrogen evolution occurring at the cathode to efficiently decline the effect of proton on the stereoselective outcomes (Fig. 9c).
This transformation was also achieved via photoredox catalysis using molecular oxygen, facilitating the formation of the electrophilic imine intermediate 68 [70]. For comparison, representative examples were selected to illustrate the differences between the two approaches (Fig. 10). In some cases, the electrochemical method afforded superior diastereoselectivity, while the photochemical method provided higher yield and enantioselectivity. Furthermore, acyclic ketones and aldehydes proved suitable under photoredox conditions, which were not reported by Mei et al. [54]. Therefore, not all electrochemical transformations surpass existing methods at this stage, thus there remains significant spaces for improvement in electrochemical enantioselective enamine catalysis.
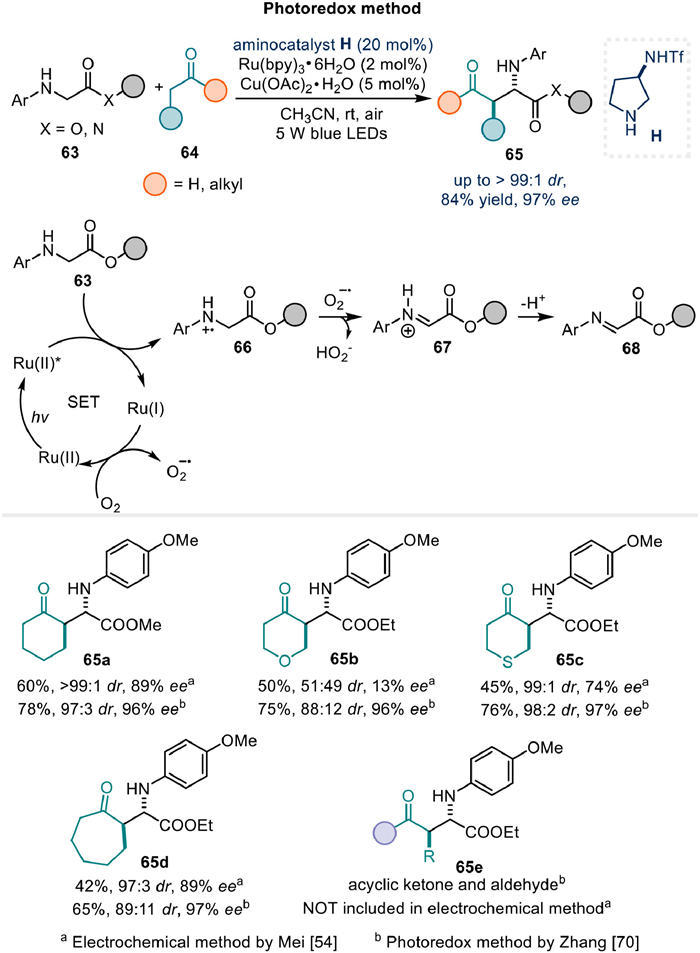
Pan's group has reported an innovative enantioselective electrochemical cross-dehydrogenative coupling reaction involving active benzylic C(sp3)-H bonds with aldehydes in 2024 (Fig. 11a) [71]. This reaction proceeds via the stereoselective capture of carbocation species generated by anodic oxidation, using enamines as nucleophiles in a polar manner. The versatility of this method is further demonstrated by the successful use of alternative coupling partners, including xanthene, methylacridine, diarylmethane, and cycloheptatriene (Fig. 11b).
The electrochemical conditions employed in this study have been well optimized to ensure superior enantioselectivity control compared to previous reports [72]. Mechanistically, taking xanthene as an example, the compound 69a exhibits a lower oxidation potential (1.15 V vs. Ag/AgCl) than both the aldehyde and the corresponding enamine intermediate, as evidenced by cyclic voltammetry studies. Moreover, a radical clock reaction using 74a, derived from a cis-cyclopropyl-substituted aldehyde exclusively yielded product 71a with a cis-substituted cyclopropane under the electrochemical conditions (Fig. 11c). This outcome further supports the proposed mechanism involving the selective oxidation of 69a to 73 via twice single electron oxidation and subsequent capture by the enamine via polar bond formation mechanism. This detailed mechanistic understanding not only validates the proposed reaction pathway but also underscores the robustness and precision of the electrochemical approach in achieving high enantioselectivity.
In a 2024 report, Xu and coworkers also brought an electrochemical cross-dehydrogenative coupling strategy for the α-alkylation of aldehydes using 9,10-dihydroacridines [73]. Distinct from Pan's work, this system employed catalytic TEMPO as a redox mediator to facilitate single-electron oxidation of dihydroacridine 76, generating radical cation intermediate 79. Subsequent reaction with O2 produced iminium ion intermediate 80, which served as the critical electrophile for stereoselective trapping by chiral enamine 81 (Fig. 12a). Mechanistic validation included control experiments with pre-synthesized 80a, confirming the intermediacy of the iminium species (Fig. 12b). Notably, the crucial C–C bond formation step was identified as a polar process.

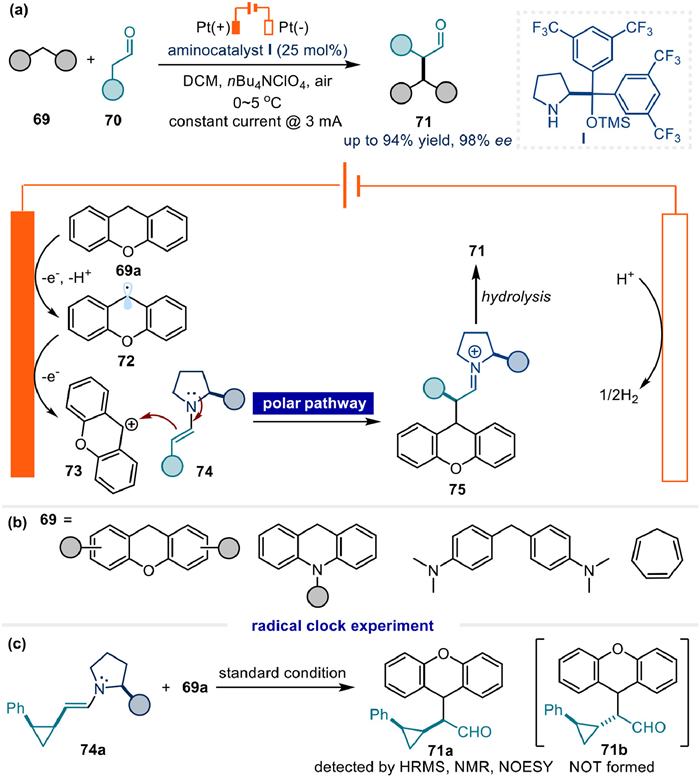
Correspondingly, chemical oxidants, such as DDQ and molecular oxygen were also compatible for this reaction as reported by Cozzi [74], Jiao [75] and Xiao (Fig. 13a) [76]. Within the photochemical transformation developed by Pericàs [77], a stoichiometric oxidant BrCCl3 was still required to generate the crucial carbocation intermediate 73 (Fig. 13b).
Meanwhile, Xu and coworkers reported an electrochemical enantioselective α-alkylation of aldehydes utilizing a newly designed bifunctional chiral aminocatalyst (Fig. 14a) [78]. The key innovation lies in the integration of a tertiary amine redox mediator moiety into a pyrrolidine-based chiral catalyst framework. This dual-functional design synergistically combines efficient electron transfer mediation with stereochemical control, providing a paradigm for achieving both high efficiency and enantioselectivity in electrochemical asymmetric catalysis.

After systematic optimization, catalyst M was proven to be the optimal from two aspects: (ⅰ) Acting as a redox mediator for the efficient oxidation of phenol 99 to radical intermediate 103 as confirmed by CV measurements. Then 103 was oxidized further at the anode to yield electrophilic para-quinone methide intermediate 104. (ⅱ) The in situ generated chiral enamine 105 from catalyst M underwent nucleophilic addition to 104, affording intermediate 106, which finally led to product 101 through hydrolysis.
Detailed reaction optimization established the optimal conditions: carbon cloth anode, platinum cathode, nBu4NPF6 electrolyte, HFIP additive in DCE at 0 ℃ under constant current (1 mA). Substituting the anode with graphite or the cathode with Ni foam afforded inferior results (Fig. 14b, entries 2 and 3). Similarly, using nBu4NClO4 as electrolyte yielded only 65% product (Fig. 14b, entry 4). The electrolyte proved essential, as its absence reduced the yield to 11% (Fig. 14b, entry 5). Importantly, HFIP was critical for high efficiency, substitutes such as CF3CH2OH, MeOH, or (CF3)2CHOCH3 significantly diminished reactivity (Fig. 14b, entries 6–8), while omission of HFIP almost suppressed product formation entirely (Fig. 14b, entry 9).
Mechanistic studies revealed that HFIP could activate the para-quinone methide via hydrogen bonding and help to improve the diastereocontrol to some extent owing to the steric hindrance. What's more, comparative studies highlighted the superiority of catalyst M over conventional systems employing separate aminocatalyst 107 and N-methylpiperidine 108, with M consistently delivering superior yields and stereoselectivity across selected substrates (Fig. 14c). This work establishes a new thinking for merging electrochemical activation with asymmetric enamine organocatalysis through bifunctional catalyst design.
Apart from above achievements utilizing classical enamine catalysis to catch electrochemical oxidation generated electrophiles, a novel SN1 type α-functionalization of aldehyde was reported by Luo's group in 2024 via α-imino carbocation intermediate (Fig. 15a) [79]. First of all, a minuscule amount of copper additive CuL was proven effective for improving the reaction rate and final yield by control experiment (Fig. 15b, entries 1 and 2). Further mechanistic studies revealed that the most probable role of copper was acting as a redox mediator for the electrochemical oxidative generation of α-imino carbocation intermediate 115.
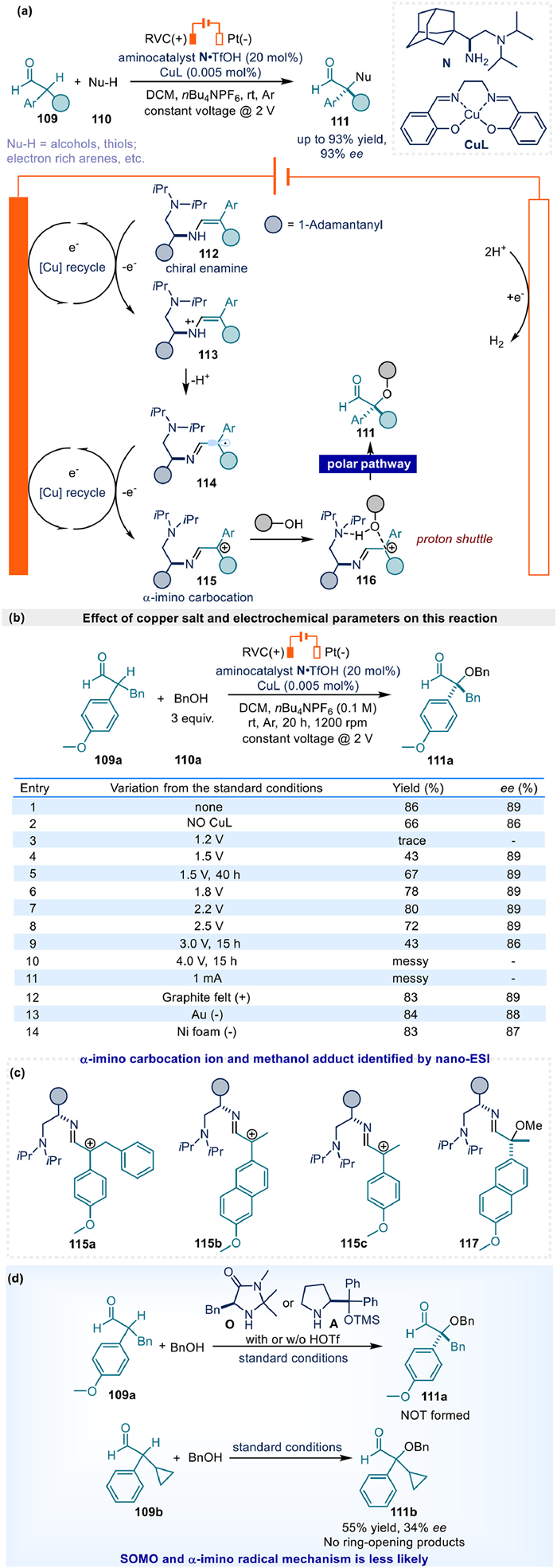
Constant voltage electrolysis proved essential for this reaction. After optimization, 2.0 V afforded optimal results (Fig. 15b, entry 1). Reactions at 1.2 V failed to generate product (Fig. 15b, entry 3), while 4.0 V produced complex mixtures with unidentified side products (Fig. 15b, entry 10). Voltages between 1.5–1.8 V and 2.2–3.0 V yielded lower efficiency (Fig. 15b, entries 4–9). Similarly, constant current electrolysis at 1 mA generated messy mixtures with unknown side products (Fig. 15b, entry 11). Attempts using graphite anodes or gold/nickel foam cathodes all afforded slightly reduced yields (Fig. 15b, entries 12–14).
Impressively, detailed mechanistic studies, including control experiments and kinetic investigations provide sufficient evidence for the formation of α-imino carbocation intermediate in the electrochemical system. For instance, α-imino carbocation intermediates 115a-115c and the methanol adduct 117 could be identified by nano-ESI, demonstrating the feasibility of proposed reaction pathway (Fig. 15c). In addition, the absence of product 111a in the reactions employing secondary chiral aminocatalysts (imidazolidinone O and prolinol A), coupled with radical clock experiments using aldehyde 109b, helpfully excluded competing SOMO and α-imino radical mechanisms (Fig. 15d).
Besides, DFT calculations provided critical insight into stereochemical control, demonstrating that the tertiary amine moiety in catalyst N orchestrates hydrogen transfer in intermediate 116 as well as the hydrogen-bonding interactions. In this scenario, the key C–C and C-X bonds formation step is also a polar process. A series of alcohols, thiols, and electron rich arenes could take part in the reaction with moderate to good yield and good enantioselectivity, while trials using N-based nucleophiles didn't work as well as others to result in 22%–24% yield and worse enantioselectivity (33%−78% ee), leaving spaces for further improvement.
Comparing to above summarized asymmetric transformations enabled by electrochemical enamine catalysis with polar chemical bond building paradigm, the radical mediated examples from radical cation or α-imino radical were still less explored.
In 2009, a pioneering reaction of α-oxyamination of aldehydes enabled by electrochemical enamine catalysis came from Jang's group (Fig. 16a) [80]. Cyclic voltammetry and control experiments indicated that electrochemical oxidation generated radical cation 122 from enamine 121 was an essential active species to react with TEMPO, yielding α-oxyaminated product 120 finally. While the C–O bond formation under the classically polar mode of enamine to undergo nucleophilic addition to TEMPO+ could be ruled out based on the outcomes that product 120a could not be formed from the reaction of enamine with commercially available TEMPO+ (Fig. 16b).
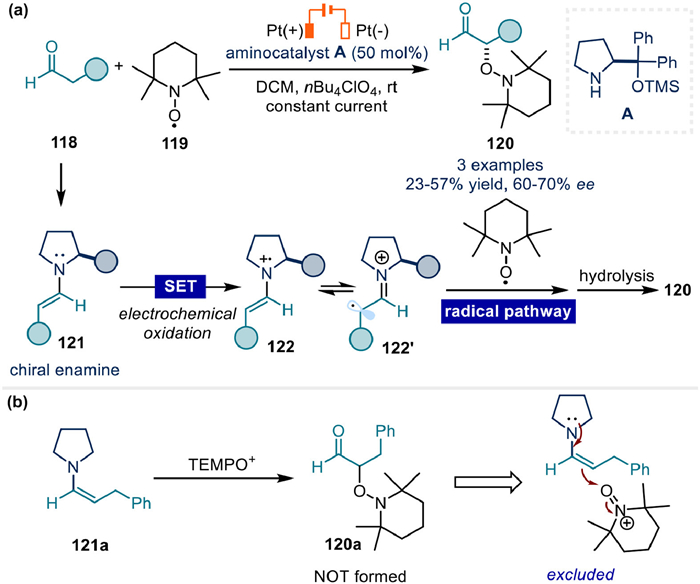
Further exploration of enantioselective variants demonstrated that 50 mol% of chiral aminocatalyst A could deliver the desired product with moderate enantioselectivity and yield. Although these stereochemical results were suboptimal, this work represented a paradigm shift by establishing the feasibility of stereoselective radical coupling with chiral enamine intermediates under electrochemical circumstance.
One year later, Jang's group reported their finding in the electrochemically driven enantioselective α-alkylation of simple aldehydes with xanthene (Fig. 17) [72]. Two mechanisms are proposed: (ⅰ) Stereoselective coupling of carbon radical 72 and α-imino radical 126′ from single electrochemical oxidation of xanthene 69a and enamine 125, respectively. In this hypothesis, the key C–C bond formation step is a radical pathway (Fig. 17, path Ⅰ). (ⅱ) Polar mode nucleophilic addition of enamine to carbocation intermediate 73 coming from anodic oxidation of 69a (Fig. 17, path Ⅱ). Mechanistic studies including CV test, adding radical trapping reagent BHT, as well as radical clock trial supported the radical coupling procedure. Finally, product 127 was formed in quite low yield and ee starting from cycloheptatriene.
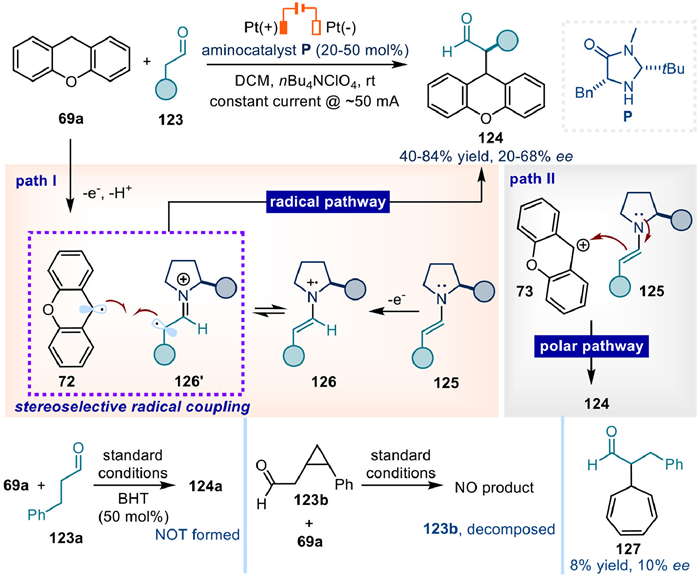
Even though the enantioselectivities were not so good (20%−68% ee) (Fig. 17), and some of which even require sub-stoichiometric amount of chiral catalyst (50 mol%), this preliminary result further demonstrated the stereoselectivity controllability under electrochemical conditions, which could work much better through continuously optimized conditions yet with different mechanism (Fig. 11) [71].
A highly enantioselective α-alkylation and allylation of aldehydes was accomplished by Dell'Amico et al. in 2024 via the electrochemical SOMO activation (Fig. 18a) [55]. Both the biphenyl additive 133 and water were quite essential for the expected high reactivity as the yield would drop significantly without their participations (Fig. 18b, entries 1–3). Furthermore, replacing the electrolyte to nBu4NBF4 or nBu4NPF6 gave 34%−61% yield compared to 70% with nBu4NClO4 (Fig. 18b, entry 4). Meanwhile, using graphite as anode was less effective compared to reticulated vitreous carbon (RVC) (Fig. 18b, entry 5).
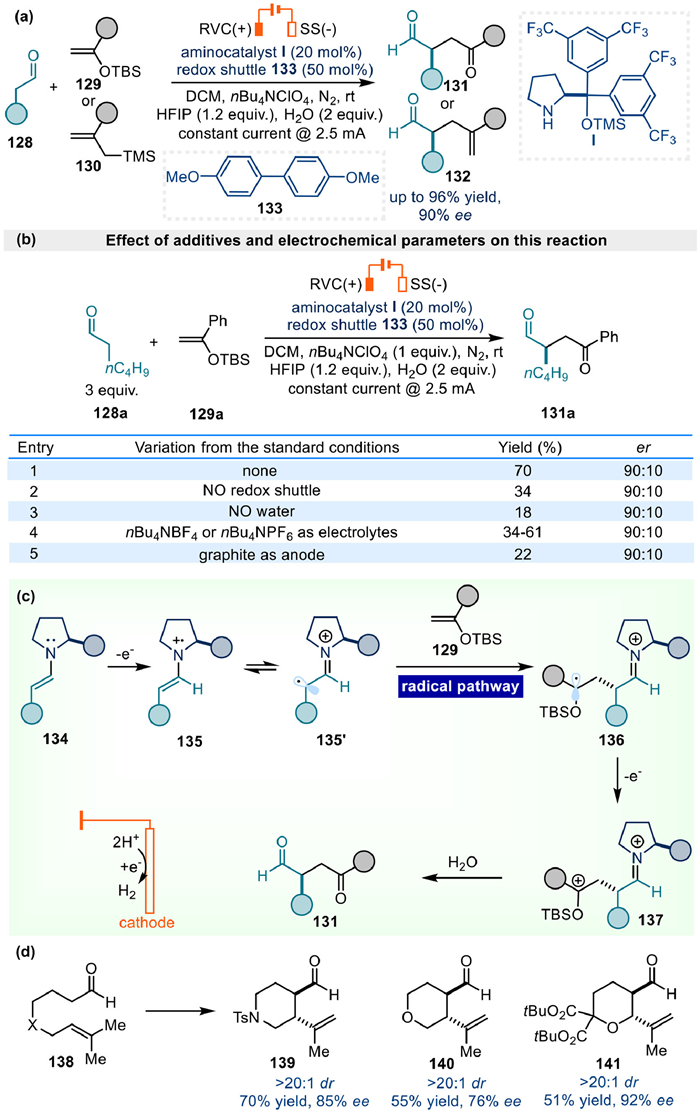
Control experiment, CV measurements, UV–vis absorption spectroscopy studies, cell potential monitoring, as well as the time-dependent density functional theory (TD-DFT) calculations indicated that a biphenyl additive 133 acted as a redox shuttle to inhibit the decomposition of chiral aminocatalyst, and to facilitate the single electron oxidation of enamine 134 to generate α-imino radical 135′ for an improved efficiency. Next, polarity match triggered reaction of 135′ with electron rich olefins, such as the silyl enol ethers, would generate intermediate 136 following a radical mediated pathway. Subsequent oxidation and hydrolysis afforded the target chiral product (Fig. 18c). Notably, intramolecular α-allylations were also accessible to afford cycloalkanes with adjacent stereocenters in high diastereoselectivity (Fig. 18d). The success of this protocol verified that an improved stereocontrol could indeed be realized by the radical mechanism under electrochemical enamine catalysis.
Recently, Luo's team successfully move the electrochemical asymmetric SOMO activation forward to develop an α-alkenylation of ketones in high enantioselectivity via synergistic enamine catalysis and electrochemistry using an organic naphthalene mediator (Fig. 19a) [81]. Based on the radical trapping results and CV measurement studies, naphthalene 145 was proven to act as an efficient mediator for the smooth oxidization of chiral enamine intermediate 147 to α-imino radical intermediate 148, ensuring an improvement of the total productivity. After that, a stereoselective C–C bond formation via radical process occurred through transition state TS III, whereas the ion-pair interaction between the protonated tertiary amine in the aminocatalyst and the anion region in the styryl trifluoroborate was identified quite important for the observed high stereoselectivity.
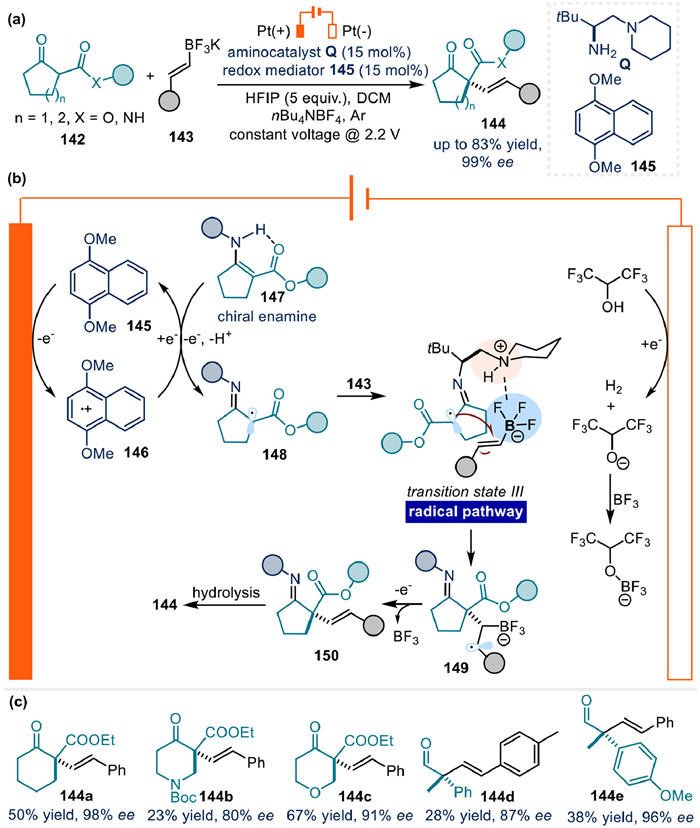
This scenario was also supported by the outcomes from stoichiometric reaction utilizing preprepared enamine with water additive, effect of boron groups, solvents, and tertiary amine structures within the aminocatalyst, as well as the DFT calculation analysis. Once formed, radical intermediate 149 would undergo a following single electron oxidation to give the expected product (Fig. 19b). Notably, except the five-membered cyclic β-ketoesters, six-membered cyclic β-ketoesters, and the ones with nitrogen and oxygen atoms, as well as acyclic aldehydes could all take part in the reaction to yield the target products in 23%–67% yields and 80%−98% ee (Fig. 19c).
In conclusion, electrochemical methodologies have emerged as a transformative platform for advancing enantioselective enamine catalysis, demonstrating dual advantages in sustainable synthesis and novel reactivity exploration. Current achievements validate their capacity to replace conventional chemical oxidants while introducing unique mechanistic dimensions, as evidenced by electricity-dependent stereochemical outcomes above. The established paradigm predominantly utilizes polar chemical bond formation through chiral enamine-mediated nucleophilic addition to electrochemically generated electrophiles. However, this technological superiority should not be misinterpreted as mere oxidant substitution, and the applied potential itself serves as an essential mechanistic variable enabling unprecedented reaction pathways.
Three critical challenges demand focused attention: (ⅰ) Oxidation sensitivity management across different components (aminocatalyst, enamine intermediate, substrates, and products), which fundamentally dictates both reaction efficiency and mechanistic feasibility; (ⅱ) Improving compatibility of classical stereocontrol strategies with complex electrochemical environments. Encouragingly, these achievements have preliminarily established reliable frameworks for asymmetric control via both polar processes (transient electrophiles) and radical mechanisms (radical species).
Future development could prioritize: (ⅰ) Expansion beyond current electro-oxidative dominance to encompass reductive radical pathways, such as the α-functionalization via electroreductively generated electrophilic radicals, as exemplified in Fig. 1B-ⅱ through photocatalytic prototypes. (ⅱ) De novo design of stereocontrol strategies leveraging electrochemical resolution. (ⅲ) Integration of advanced characterization techniques for real-time mechanistic elucidation.
The demonstrated successes, while significant, merely outline the preliminary chapter in what promises to be a transformative approach to asymmetric synthesis. We anticipate that the synergistic integration of electrochemical evolution with enamine catalysis will drive the emergence of innovative catalytic paradigms, potentially revolutionizing modern synthetic methodologies through unprecedented reaction pathways and selectivity control.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Dengke Ma: Writing – review & editing, Writing – original draft, Visualization, Validation, Funding acquisition, Conceptualization. Youai Qiu: Writing – review & editing, Validation, Conceptualization.
Financial support from National Natural Science Foundation of China (No. 22201187), and the starting grant from Capital Medical University (No. 3100–12500121) are gratefully acknowledged.
C.C.C. Johansson, T.J. Colacot, Angew. Chem. Int. Ed. 49 (2010) 676–707. doi: 10.1002/anie.200903424
A.M.R. Smith, K.K. Hii, Chem. Rev. 111 (2011) 1637–1656. doi: 10.1021/cr100197z
S. Liang, K. Xu, C.C. Zeng, et al., Adv. Synth. Catal. 360 (2018) 4266–4292. doi: 10.1002/adsc.201800519
L. Zhu, D. Wang, Z. Jia, et al., ACS. Catal. 8 (2018) 5466–5484. doi: 10.1021/acscatal.8b01263
B. List, Acc. Chem. Res. 37 (2004), 548–557. doi: 10.1021/ar0300571
W. Notz, F. Tanaka, C.F. Barbas, Acc. Chem. Res. 37 (2004) 580–591. doi: 10.1021/ar0300468
S. Mukherjee, J.W. Yang, S. Hoffmann, et al., Chem. Rev. 107 (2007) 5471–5569. doi: 10.1021/cr0684016
P. Melchiorre, M. Marigo, A. Carlone, et al., Angew. Chem. Int. Ed. 47 (2008) 6138–6171. doi: 10.1002/anie.200705523
L. Xu, J. Luo, Y. Lu, Chem. Commun. 2009 (2009) 1807–1821. doi: 10.1039/b821070e
S. Bertelsen, K.A. Jørgensen, Chem. Soc. Rev. 38 (2009) 2178–2189. doi: 10.1039/b903816g
A. Quintard, S. Belot, E. Marchal, et al., Eur. J. Org. Chem. 2010 (2010) 927–936. doi: 10.1002/ejoc.200901283
M. Nielsen, D. Worgull, T. Zweifel, et al., Chem. Commun. 47 (2011) 632–649. doi: 10.1039/C0CC02417A
K.L. Jensen, G. Dickmeiss, H. Jiang, et al., Acc. Chem. Res. 45 (2012) 248–264. doi: 10.1021/ar200149w
T. Kano, K. Maruoka, Chem. Sci. 4 (2013) 907–915. doi: 10.1039/C2SC21612D
B.M. Paz, H. Jiang, K.A. Jorgensen, Chem. Eur. J. 21 (2015) 1846–1853. doi: 10.1002/chem.201405038
S. Meninno, C. Volpe, A. Lattanzi, ChemCatChem. 11 (2019) 3716–3729. doi: 10.1002/cctc.201900569
T.D. Beeson, A. Mastracchio, J. -B. Hong, et al., Science 316 (2007) 582–585. doi: 10.1126/science.1142696
M. Mečiarová, P. Tisovský, R. Šebesta, New J. Chem. 40 (2016) 4855–4864. doi: 10.1039/C6NJ00079G
Y.Q. Zou, F.M. Hörmann, T. Bach, Chem. Soc. Rev. 47 (2018) 278–290. doi: 10.1039/c7cs00509a
D. Ma, T.H.F. Wong, P. Melchiorre, Asymmetric photoredox reactions without photocatalysts, in: T. Akiyama, I. Ojima (Eds.), Catalytic Asymmetric Synthesis, 4th ed., John Wiley & Sons, Inc., Hoboken, 2022, pp. 329–354.
D.A. Nicewicz, D.W.C. MacMillan, Science 322 (2008) 77–80. doi: 10.1126/science.1161976
D.A. Nagib, M.E. Scott, D.W.C. MacMillan, J. Am. Chem. Soc. 131 (2009) 10875–10877. doi: 10.1021/ja9053338
H.W. Shih, M.N. Vander Wal, R.L. Grange, et al., J. Am. Chem. Soc. 132 (2010) 13600–13603. doi: 10.1021/ja106593m
M. Neumann, S. Füldner, B. König, et al., Angew. Chem. Int. Ed. 50 (2011) 951–954. doi: 10.1002/anie.201002992
Y. Zhu, L. Zhang, S. Luo, J. Am. Chem. Soc. 136 (2014) 14642–14645. doi: 10.1021/ja508605a
E. Arceo, I.D. Jurberg, A. Álvarez-Fernández, et al., Nat. Chem. 5 (2013) 750–756. doi: 10.1038/nchem.1727
M. Silvi, E. Arceo, I.D. Jurberg, et al., J. Am. Chem. Soc. 137 (2015) 6120–6123. doi: 10.1021/jacs.5b01662
A. Bahamonde, P. Melchiorre, J. Am. Chem. Soc. 138 (2016) 8019–8030. doi: 10.1021/jacs.6b04871
G. Filippini, M. Silvi, P. Melchiorre, Angew. Chem. Int. Ed. 56 (2017) 4447–4451. doi: 10.1002/anie.201612045
E.J. Horn, B.R. Rosen, P.S. Baran, ACS Cent. Sci. 2 (2016) 302–308. doi: 10.1021/acscentsci.6b00091
M. Yan, Y. Kawamata, P.S. Baran, Chem. Rev. 117 (2017) 13230–13319. doi: 10.1021/acs.chemrev.7b00397
Y. Jiang, K. Xu, C. Zeng, Chem. Rev. 118 (2018) 4485–4540. doi: 10.1021/acs.chemrev.7b00271
J. -i. Yoshida, A. Shimizu, R. Hayashi, Chem. Rev. 118 (2018) 4702–4730. doi: 10.1021/acs.chemrev.7b00475
T.H. Meyer, I. Choi, C. Tian, et al., Chem. 6 (2020) 2484–2496. doi: 10.1016/j.chempr.2020.08.025
C. Zhu, N.W.J. Ang, T.H. Meyer, et al., ACS Cent. Sci. 7 (2021) 415–431. doi: 10.1021/acscentsci.0c01532
L.F.T. Novaes, J.J. Liu, Y.F. Shen, et al., Chem. Soc. Rev. 50 (2021) 7941–8002. doi: 10.1039/d1cs00223f
X. Cheng, A.W. Lei, T. -S. Mei, et al., CCS Chem. 4 (2022) 1120–1152. doi: 10.31635/ccschem.021.202101451
Q.L. Zhang, K. Liang, C. Guo, Sci. China Chem. 67 (2024) 755–758. doi: 10.1007/s11426-023-1832-7
W. Zeng, Y. Wang, C. Peng, et al., Chem. Soc. Rev. 54 (2025) 4468–4501. doi: 10.1039/d4cs01142b
J. Zhong, Y. Yu, D. Zhang, et al., Chin. Chem. Lett. 32 (2021) 963–972. doi: 10.1016/j.cclet.2020.08.011
W. Zhou, X. Chen, L. Lu, et al., Chin. Chem. Lett. 35 (2024) 08902.
P.Y. Chen, C. Huang, L.H. Jie, et al., J. Am. Chem. Soc. 146 (2024) 7178–7184. doi: 10.1021/jacs.4c00878
P. Zhou, T. Zou, H.J. Song, et al., Chin. Chem. Lett. 37 (2026) 111673. doi: 10.1016/j.cclet.2025.111673
S. Rani, N. Sbei, S. Rahali, et al., Chin. Chem. Lett. 36 (2025) 111216. doi: 10.1016/j.cclet.2025.111216
Q. Lin, L. Li, S. Luo, Chem. Eur. J. 25 (2019) 10033–10044. doi: 10.1002/chem.201901284
M. Ghosh, V.S. Shinde, M. Rueping, Beilstein J. Org. Chem. 15 (2019) 2710–2746. doi: 10.3762/bjoc.15.264
C. Margarita, H. Lundberg, Catalysts. 10 (2020) 982. doi: 10.3390/catal10090982
X. Chang, Q. Zhang, C. Guo, Angew. Chem. Int. Ed. 59 (2020) 12612–12622. doi: 10.1002/anie.202000016
K.J. Jiao, Z.H. Wang, C. Ma, et al., Chem. Catal. 2 (2022) 3019–3047.
F. Medici, S. Resta, S. Andolina, et al., Catalysts. 13 (2023) 944. doi: 10.3390/catal13060944
J. Rein, S.B. Zacate, K. Mao, et al., Chem. Soc. Rev. 52 (2023) 8106–8125. doi: 10.1039/d3cs00511a
C. Gao, X. Liu, M. Wang, et al., Chin. J. Org. Chem. 44 (2024) 673–727. doi: 10.6023/cjoc202402005
X. Jiang, C. Zou, W. Zhuang, et al., Green. Chem. 27 (2025) 915–945. doi: 10.1039/d4gc05316h
Z. -H. Wang, P. -S. Gao, X. Wang, et al., J. Am. Chem. Soc. 143 (2021) 15599–15605. doi: 10.1021/jacs.1c08671
D. Mazzarella, C. Qi, M. Vanzella, et al., Angew. Chem. Int. Ed. 63 (2024) e202401361. doi: 10.1002/anie.202401361
K.L. Jensen, P.T. Franke, L.T. Nielsen, et al., Angew. Chem. Int. Ed. 49 (2010) 129–133. doi: 10.1002/anie.200904754
N. Fu, L. Li, Q. Yang, et al., Org. Lett. 19 (2017) 2122–2125. doi: 10.1021/acs.orglett.7b00746
X. Liu, S. Sun, Z. Meng, et al., Org. Lett. 17 (2015) 2396–2399. doi: 10.1021/acs.orglett.5b00909
X. Liu, Z. Meng, C. Li, et al., Angew. Chem. Int. Ed. 54 (2015) 6012–6015. doi: 10.1002/anie.201500703
G. Zhang, Y. Ma, S. Wang, et al., Chem. Sci. 4 (2013) 2645–2651. doi: 10.1039/c3sc50604e
Q. Yang, L. Zhang, C. Ye, et al., Angew. Chem. Int. Ed. 56 (2017) 3694–3698. doi: 10.1002/anie.201700572
H. Hou, S. Zhu, L. Atodiresei, et al., Eur. J. Org. Chem. 2018 (2018) 1277–1280. doi: 10.1002/ejoc.201800117
F.Y. Lu, Y.J. Chen, Y. Chen, et al., Chem. Commun. 56 (2020) 623–626. doi: 10.1039/c9cc09178e
X. Liu, X. Yan, J.H. Yu, et al., Org. Lett. 21 (2019) 5626–5629. doi: 10.1021/acs.orglett.9b01965
C.L. Dong, X. Ding, L.Q. Huang, et al., Org. Lett. 22 (2020) 1076–1080. doi: 10.1021/acs.orglett.9b04613
L. Li, Y. Li, N. Fu, et al., Angew. Chem. Int. Ed. 59 (2020) 14347–14351. doi: 10.1002/anie.202006016
R. Francke, R.D. Little, Chem. Soc. Rev. 43 (2014) 2492–2521. doi: 10.1039/c3cs60464k
J.E. Nutting, M. Rafiee, S.S. Stahl, Chem. Rev. 118 (2018) 4834–4885. doi: 10.1021/acs.chemrev.7b00763
F. Wang, S.S. Stahl, Acc. Chem. Res. 53 (2020) 561–574. doi: 10.1021/acs.accounts.9b00544
X. Yang, Z. Xie, Y. Li, et al., Chem. Sci. 11 (2020) 4741–4746. doi: 10.1039/d0sc00683a
Y. Chen, Y. Wu, G. Wang, et al., Chin. Chem. Lett. 35 (2024) 109445. doi: 10.1016/j.cclet.2023.109445
X.H. Ho, S.I. Mho, H. Kang, et al., Eur. J. Org. Chem. 2010 (2010) 4436–4441. doi: 10.1002/ejoc.201000453
J.Y. He, N.N. Wang, R. Zhao, et al., Eur. J. Org. Chem. 27 (2024) e202400817. doi: 10.1002/ejoc.202400817
F. Benfatti, M.G. Capdevila, L. Zoli, et al., Chem. Commun. 2009 (2009) 5919–5921. doi: 10.1039/b910185c
B. Zhang, S.K. Xiang, L.H. Zhang, et al., Org. Lett. 13 (2011) 5212–5215. doi: 10.1021/ol202090a
F. Huang, L. Xu, J. Xiao, Chin. J. Chem. 30 (2012) 2721–2725. doi: 10.1002/cjoc.201200850
E. Larionov, M.M. Mastandrea, M.A. Pericàs, ACS. Catal. 7 (2017) 7008–7013. doi: 10.1021/acscatal.7b02659
J.Y. He, C. Zhu, W.X. Duan, et al., Angew. Chem. Int. Ed. 63 (2024) e202401355. doi: 10.1002/anie.202401355
Q. Lin, Y. Duan, Y. Li, et al., Nat. Commun. 15 (2024) 6900. doi: 10.1038/s41467-024-50945-2
N.N. Bui, X.H. Ho, S.I. Mho, et al., Eur. J. Org. Chem. 2009 (2009) 5309–5312. doi: 10.1002/ejoc.200900871
Y. Duan, Y. Zhang, J. Chen, et al., Angew. Chem. Int. Ed. (2025) e202505826. doi: 10.1002/anie.202505826

Figure 2 Mechanistic distinctions between polar and radical-mediated pathways in electrochemical enantioselective enamine catalysis.

Figure 3 Electrochemical enantioselective synthesis of meta-alkylated anilines via organocatalytic α-arylation of enamine.

Figure 4 Electrochemical oxidative cross-dehydrogenative coupling of tetrahydroisoquinoline and simple ketones.

Figure 5 Enantioselective cross-dehydrogenative coupling of tetrahydroisoquinoline and simple ketones enabled by thermodynamic and photoredox conditions.

Figure 6 Diastereo- and enantio–selective electrosynthesis of indolin-3-ones with 2-quaternary stereocenters.

Figure 7 Diastereo- and enantio–selective synthesis of indolin-3-ones with 2-quaternary stereocenters enabled by thermodynamic and photoredox conditions.

Figure 8 Electrochemically enabled asymmetric α-arylation of cyclic β-ketocarbonyl compounds via enamine catalysis.

Figure 9 TEMPO mediated electrochemical stereoselective Shono-type cross dehydrogenative coupling of acyclic amines and cyclic ketones.

Figure 10 Photoredox cross-dehydrogenative coupling of glycine derivatives with ketones and aldehydes.

Figure 11 Enantioselective electrochemical cross-dehydrogenative coupling of aldehydes and benzylic C(sp3)-H bonds.

Figure 12 Enantioselective electrochemical α-alkylation of aldehydes using 9,10-dihydroacridines.

Figure 13 Enantioselective dehydrogenative α-alkylation of aldehydes enabled by thermodynamic and photoredox conditions.

Figure 14 Electrochemical enantioselective α-alkylation of aldehydes using bifunctional chiral aminocatalyst.

Figure 15 Electrochemical enantioselective α-functionalization of aldehydes via α-imino carbocation intermediate.

Figure 16 Electrochemical α-oxyamination of aldehydes via enamine catalysis and preliminary enantioselective results.

Figure 17 Electrochemically driven enantioselective α-alkylation of simple aldehydes with xanthene.

Figure 18 Radical based electrochemical enantioselective α-alkylation and allylation of aldehydes.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:

 下载:
下载: