

Figure 1.
TGA curves for the SiO2, initiator-functionalized silica particles, lithium polystyrene sulfonate brushes grafted silica particles.
Enhancing the ionic conductivity in a composite polymer electrolyte with ceramic nanoparticles anchored to charged polymer brushes
Bintao Zhao , Xi Lu , Qian Wang , Jingfa Yang , Jiang Zhao , Henghui Zhou

Lithium-ion batteries have been used in many areas in recent years, including portable, vehicle and consumable electronic products [1]. However, due to the use of a large amount of organic electrolyte and the presence of flammable carbonated solvents, conventional liquid lithium batteries have the potential to catch on fire or explode at high temperatures. These defects are the main obstacles restricting their application [2, 3]. Recently, considerable effort has been dedicated to improving the performance of current rechargeable lithium batteries due to the increasing demand of safe lithium batteries [3, 4]. It is urgent to produce very safe lithium batteries to satisfy these demands.
An interesting research direction is solid lithium batteries. A solid-state ion battery is a new way to avoid the use of organic electrolytes and obtain an acceptable safety and a high energy density [5-7]. In addition, solid electrolytes also have the potential to suppress lithium metal dendrites because of their high mechanical strength and large electrochemical stability windows. All these advantages suggest that solid batteries have a bright future in practice. However, the low ionic conductivity compared with liquid lithium batteries is the main obstacle for solid electrolytes. Although researchers are indeed making a considerable effort, this defect is still the main material challenge in solid batteries [8].
Two general strategies are adopted to produce solid electrolytes, including inorganic materials and organic polymer materials [9-11]. However, the low compatibility between inorganic materials and the electrode, which causes a high interfacial resistance, restricts its practical application [12]. In contrast, polymer electrolytes possess flexibility, light weight and good compatibility with electrodes and have attracted increasing attention in recent years [5, 13, 14]. Several types of solid polymer electrolytes such as poly(ethylene oxide) (PEO), polyacrylonitrile (PAN), poly(vinylidene fluoride) and polyester, have been extensively studied [15-18]. However, the low ionic conductivity, poor electrochemical stability, and low mechanical strength, which may form a short circuit during long cycling because of lithium dendrites, restrict their application in commercial lithium batteries. In order to solve these problems, a variety of approaches, including crosslinking between different polymer chains, or synthesis of new block polymers [19], have been developed. Although these strategies increase the mechanical strength of polymer electrolytes, these methods deteriorate the compatibility with electrodes and decrease the ionic conductivity. In contrast, adding ceramic nanoparticles can simultaneously increase the ionic conductivity and mechanical strength, which improves the ability to suppress lithium dendrites [20, 21]. Since the first publication about dispersing ceramic particles into a polymer electrolyte by Weston [22], researchers have reached a consensus about ceramic nanoparticles increasing the ionic conductivity. Dispersing ceramic nanoparticles hinders polymer crystallization and provides a highly conductive interface between the nanoparticles and polymer electrolyte [18, 23-25]. However, nanoparticles have the potential to aggregate in the polymer electrolyte, damaging the performance of lithium batteries. To solve this problem, surface groups are grafted to the nanoparticles to improve their dispersing performance, further increasing the electrochemical properties of composite polymer electrolytes. This phenomenon is ascribed to the fast throughput of the conducting pathways at the surface layers [23]. However, all these publications are concern about small molecules anchored to the nanoparticles. Few researchers pay attention to the nanoparticles which are modified by the long charged polymer chains, which have the ability to dissociate the free ions. Charged anion monomers can increase the lithium-ion transference number and elevate the ionic conductivity of the polymer composite [26-28]. The work by Lee has proved that core-shell structure is useful in gel polymer electrolyte battery. If this strategy is a promising way in solid lithium battery, it still requires us to explore.
In the current work, we synthesized spherical polymer brushes containing core-shell structures. Polymer brushes [29], consisting of polymers chains dense grafted onto interfaces by chemical bonding, have the ability to combine the properties of charged polymer chains and substrates. Through facile in situ polymerization, poly(vinylene carbonate) (PVC) and a spherical SiO2 polymer brush electrolyte (SSP) are generated. The sulfonate group is chemically anchored to the polymer chains and has the high ability to dissociate mobile cations [30]. Owing to the charged polymer brushes outside the ceramic particles, SSP polymer electrolytes have a higher ionic conductivity than that of the SiO2 polymer electrolytes and ceramic free PVC polymer electrolytes. This modified composite polymer electrolyte also maintains a high lithium ion transference number and excellent cycling performance, which may be a promising strategy to produce high-performance polymer electrolytes for commercial lithium batteries.
In order to acquire the composite polymer electrolyte, sphere polymer brushes are synthesized [31]. The size of pure SiO2 is about 100 nm and more details are supplied in the Supporting information.
SiO2-LiPSS particles (1 g) were ultrasonically dispersed in 2 mL ethanol. A total of 270 μL SiO2-LiPSS particles (10 wt%) solution and 1 mg of AIBN were added to 1 mL of vinylene carbonate and dried under vacuum at 50 ℃ for 24 h to remove ethanol. Then, 287 mg LiTFSI (1 mol/L LiTFSI in VC) was added. The solution was mixed thoroughly and 35 μL of the solution was injected into a 2032 lithium battery, where a 30 μm cellulose separator, purchased from NKK company, was adopted as a supporting layer for the polymer electrolyte, and separated the cathode and anode. Later, the 2032 batteries were stored at 65 ℃ for 24 h. Pure VC solution and SiO2 nanoparticles added into the VC solutions were used for comparation.
The ionic conductivities of polymer electrolyte were determined using AC impedance. Linear sweep voltammetry of polymer electrolyte was tested on a Li/polymer electrolyte/stainless steel cells at a scanning rate of 1 mV/s and the lithium ion transfer number of polymer electrolyte was calculated using equation:
|
|
where V was the applied voltage, I0 and R0 were initial current and resistance before polarization respectively. ISS and RSS were state current and resistance after polarization respectively.
For cell performance measurement, the LiFePO4 cathode was used by mixing 80 wt% LiFePO4, 10 wt% acetylene black and 10 wt% PVDF coated on an aluminum foil. The cycling performance of LiFePO4/polymer electrolyte/Li were measured using the Land battery test system between 2.5 V and 4.0 V at 25 ℃.
In our experiment, the SiO2 nanoparticles are immersed in ethanol solution with an initiator, and the FTIR spectra are used to confirm the structure of the SiO2 nanoparticles. The data are shown in Fig. S2 (Supporting information). The characteristic absorption peaks at 2920 cm-1 and 2845 cm-1 are attributed to the stretching vibration of bromine atoms, proving that the initiators have been successfully anchored to the surface of the SiO2. Because the absorption peak between the sulfonate group and the Si-O bond have overlapped, thermogravimetric analysis (TGA) are used to determine whether chains have been anchored to the nanoparticles. The polymer chains grafted to the nanoparticles could be distinguished from the difference in weight loss between the original silica particles and the charge polymer brushes grafted to the silica particles. The difference shown in Fig. 1 demonstrates that the polymer chains exist on the surface of the SiO2. The weight loss is small even if the temperature increases to 500 ℃, demonstrating that these charged polymer chains have the capacity to resist high temperatures, which may be useful in high temperature batteries. The mass of polymer chains anchored to the nanoparticles are measured by TGA and data are showed in Fig. S3 (Supporting information).
Liquid VC had been proved that these monomers could be polymerized into PVC by an initialized radical initiator for 24 h [32]. Fourier transform infrared spectroscopy is used to analyze the structure of the PVC as shown in Fig. S4 (Supporting information), which shows that a new peak appeared at 2976 cm-1, which could be ascribed to the change from a C=C bond to a C-C bond. Owing to the oxygen atom on the polymer chains, the lithium salt, LiTFSI, could be dissolved in the polymer electrolyte.
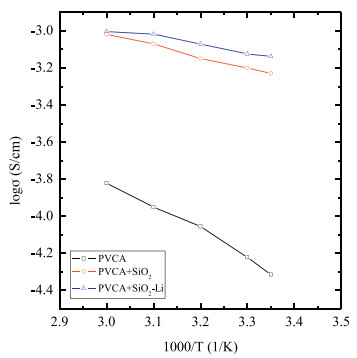
To explore the properties of the SSP composite polymer electrolyte, PVC polymer electrolyte and pure SiO2 composite polymer electrolyte are included for comparison. Fig. 2 shows the ionic conductivity of these three polymer electrolytes. The ionic conductivity of the pure PVC polymer electrolyte is approximately 5×10-5 S/cm at 25 ℃. Adding SiO2 nanoparticles increase the ionic conductivity drastically to 5×10-4 S/cm at 25 ℃. Ceramic nanoparticles decrease the ordered structure of the polymer electrolyte and provide fast movement throughout the conductive pathways at the ceramic extended surface for lithium ion [22, 33]. Compared with that of the pure SiO2 particles, the SiO2 particles with polymer brushes on the outside further increase the ionic conductivity at ambient temperature. Nanoparticles assembled with charged polymer chains are proved to increase the ionic conductivity when coated on the polyethylene separator and lithium ionic battery [28, 34]. In a polymer electrolyte system, charged chains anchored to the nanoparticles could be thought of as single ion conductors, and charged monomers could decrease the movement of anions and suppress the concentration gradients [27, 35]. Beside, Croce [23] explore the effect of different surface interaction and found that acidic monomers increase the free Li+ inducing fast throughout the conducting pathways at the ceramic extendend surface. The specific interactions exist between the anion and Li+. In addition, due to their high dissociation ability to yield mobile cations of charged polymer chains [26, 30], lithium ions transfer faster through the polymer layers and induce a high ionic conductivity compared with that of pure SiO2 particles. The ionic conductivity of the SSP composite polymer electrolyte reaches 7.3×10-4 S/cm at 25 ℃ and reaches 10-3 S/cm at 60 ℃, which is close to the performance of a liquid lithium ion battery, meaning composites with modified nanoparticles might be a practical route for solid commercial batteries.
In lithium ion batteries, only lithium ions participate in the chemical reaction and the lithium ion transference number (tLi+) is an essential factor for the enhanced performance of lithium batteries. The lithium ion transference number is calculated by the Bruce-Vincent-Evans equation. The dates are showed in Fig. 3. The PVC polymer electrolyte have a low glass transition temperature, the lithium ions shuttle easily between the cathode and anode. The equivalent circuit of AC impedance behavior is inserted in the graph. The tLi+ calculated from the equation is 0.53 and the data are shown in Fig. 3a. When SiO2 particles are added, the lithium transference number improves due to surface interaction between the nanoparticles and polymer electrolyte [24]. The pure SiO2 particles increase the tLi+ to 0.62. When exchanged with SiO2-LiPSS particles, the tLi+ continues to increase to 0.68. In addition to the ceramic nanoparticles promoting the relaxation of the polymer chains, which enhanced the mobility of the lithium ions, the charged polymer chains contain a large number of mobile lithium ions, and the cations hop easily on the surface of the polymer layers. In this way, the lithium ion transference number is higher than that of the SiO2 composite polymer electrolyte.

The electrochemical stability window of the polymer electrolytes is essential for their applications in commercial lithium batteries. Fig. S5 (Supporting information) shows the results of linear sweep voltammetry of the polymer electrolyte when scanning from 0 to 5.5 V (vs. Li+/Li). The results show that the PVC electrolyte is stable before the potential reached 4.5 V. Compared with that of the SiO2-PVC composite polymer electro-lyte and the SSP composite polymer electrolyte, the electrochemical stability window increase, and the current density begin to increase at 5 V for the SSP composite polymer electrolyte. The current density changes implied the oxidative decomposition of the composite polymer electrolyte. This voltage also confirms that the SSP composite polymer electrolyte may be a candidate material for high voltage lithium batteries.
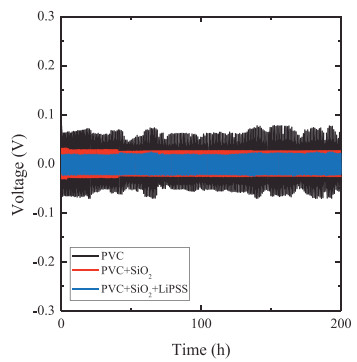
The compatibility between the electrolyte and electrode is essential for the long cycling performance. Li/polymer electrolyte/ Li batteries are assembled in the glove box filled with an Ar atmosphere. The long-term lithium deposition/striping cycles are measured at a current density of 0.1 mA/cm.
Fig. 4 shows that there are no obvious short circuits and the voltages are stable for 200 h. The results prove that the interface between the polymer electrolyte and electrode is stable. The overpotential of the SSP composite polymer electrolyte (20 mV) is smaller than that of the others. This could be ascribed to the low resistance of the batteries. The stability of the voltage during the long cycling reveals that the composite polymer electrolytes have sufficient rigidity to suppress lithium dendrites.
The electrochemical performance of these three different cells are compared to demonstrate the effectiveness of the core-shell structures of the nanoparticles dispersed in the polymer electrolytes. Adding SiO2 nanoparticles also improve the performance of cells compared to that of the pure PVC polymer electrolyte. Nanoparticles could decrease the crystallinity of the polymer electrolyte [36], so the amount of amorphous structure increase, increasing the paths for the transfer of lithium ions. Compared with that of the pure SiO2 particles, the polymer chains anchored to the surface of the nanoparticles provide a rapid transport for lithium ions at the interface. For these reasons, the rate capacity and cycling performance of the SSP composite polymer electrolyte at different current densities are improved and the dates are shown in Figs. 5a and b. The current density increases gradually from 0.1C to 1C for 5 cycles. The SSP composite polymer electrolyte has a better discharge capacity and retains 95% at 1 C after 100 cycles, corresponding to 70% of the PVC polymer electrolyte system compared with the discharge capacity of the first cycle at 1C. The pure PVC polymer composite retains stability of approximately only 60 cycles, and the discharge capacity of the cells decreases rapidly. Fig. 5c shows the charge/discharge curves of the SSP composite polymer electrolyte for the 1st, 50th and 100th cycles. The curves demonstrate that the plateaus of charge/discharge are the same and that the capacity is nearly equal. The interface between cathode and polymer electrolyte are investigated after 100 cycles. SEM images (Fig. S6 in Supporting information) show that polymer electrolyte adhere to the cathode. These data prove that the SSP composite polymer electrolyte is stable without an increasing polarization. The cycle performance also proves that nanoparticles anchored charged polymer chains, have a positive effect on increasing the performance of cells. All these results demonstrate that the SSP composite polymer electrolytes have a better electrochemical performance including the rate and cycling performance, than the SiO2 composite polymer electrolytes and PVC polymer electrolytes.

In our experiments, composite SSP polymer electrodes are synthesized and compare with composite SiO2 polymer electrolytes and PVC electrolytes. Charged polymer chains anchored to the nanoparticles show an excellent ionic conductivity and long cycling performance. The ionic conductivity can be improved to 5×10-4 S/cm at 25 ℃, and 90% long cycling retention is achieved. Owing to the polymer chains that grafted to the SiO2 particles, the lithium ions can transport very quickly in the brush layers at the interface and increase the lithium-ion transference number. These results demonstrate that the development of composite modified nanoparticles may be a promising way to facilitate the composite polymer electrolyte.
The authors thank Pulead Technology Industry Co., Ltd. for applying cathode active materials LiFePO4 for this work. This work is financially supported by PULEAD Technology Industry Co., Ltd., the National Natural Science Foundation of China (Nos. 21771018 and 21875004).
Supplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.06.009.
Y.G. Guo, J.S. Hu, L.J. Wan, Adv. Mater. 20(2008) 2878-2887. doi: 10.1002/adma.200800627
K. Xu, Chem. Rev. 104(2004) 4303-4418. doi: 10.1021/cr030203g
S.H. Kim, K.H. Choi, S.J. Cho, E.H. Kil, S.Y. Lee, J. Mater. Chem. A 1(2013) 4949-4955. doi: 10.1039/c3ta10612h
G.Y. Zheng, L. Seok Woo, L. Zheng, et al., Nat. Nanotechnol. 9(2014) 618-623. doi: 10.1038/nnano.2014.152
W. He, Z. Cui, X. Liu, et al., Electrochim. Acta 225(2017) 151-159. doi: 10.1016/j.electacta.2016.12.113
J.J. Zhang, J.H. Zhao, L.P. Yue, et al., Adv. Energy Mater. 5(2015) 1501082. doi: 10.1002/aenm.201501082
Y. Shao, H. Wang, Z. Gong, D. Wang, L. Chen, ACS Energy Lett. 3(2018) 1212-1218. doi: 10.1021/acsenergylett.8b00453
J.F.M. Oudenhoven, L. Baggetto, P.H.L. Notten, Adv. Energy Mater. 1(2011) 10-33. doi: 10.1002/aenm.201000002
Y.H. Zhu, J. Cao, L.H. Chen, et al., J. Mater. Chem. A 7(2019) 6832-6839. doi: 10.1039/C9TA00560A
Z. Lin, X. Guo, H. Yu, Nano Energy 41(2017) 646-653. doi: 10.1016/j.nanoen.2017.10.021
M. Mushtaq, X.W. Guo, J.P. Bi, Z.X. Wang, H.J. Yu, Rare Met. 37(2018) 520-526. doi: 10.1007/s12598-018-1044-8
H.L. Wan, G. Peng, X.Y. Yao, et al., Energy Storage Mater. 4(2016) 59-65. doi: 10.1016/j.ensm.2016.02.004
S.Z. Zhang, X.H. Xia, D. Xie, et al., J. Power Sources 409(2019) 31-37. doi: 10.1016/j.jpowsour.2018.10.088
Y. Cui, J. Chai, H. Du, et al., J. ACS Appl. Mater. Interfaces 9(2017) 8737-8741. doi: 10.1021/acsami.6b16218
D.M. Smith, B. Dong, R.W. Marron, et al., Nano Lett. 12(2012) 310-314. doi: 10.1021/nl203599y
Y. Xia, Y.F. Liang, D. Xie, et al., Chem. Eng. J. 358(2019) 1047-1053. doi: 10.1016/j.cej.2018.10.092
X. Pan, T. Liu, D.J. Kautz, et al., J. Power Sources 403(2018) 127-136. doi: 10.1016/j.jpowsour.2018.09.080
W.D. Zhou, S.F. Wang, Y.T. Li, et al., J. Am. Chem. Soc. 138(2016) 9385-9388. doi: 10.1021/jacs.6b05341
R. Khurana, J.L. Schaefer, L.A. Archer, G.W. Coates, J. Am. Chem. Soc.136(2014) 7395-7402. doi: 10.1021/ja502133j
F. Croce, R. Curini, A. Martinelli, et al., J. Phys.Chem. B 103(1999) 10632-10638. doi: 10.1021/jp992307u
Z. Zhang, Q. Zhang, R. Cheng, et al., J. Mater. Chem. A 4(2016) 15823-15828. doi: 10.1039/C6TA07590H
J.E. Weston, B.C.H. Steele, Solid State Ionics 7(1982) 75-79. doi: 10.1016/0167-2738(82)90072-8
F. Croce, L. Persi, B. Scrosati, et al., J. Electrochim. Acta 46(2001) 2457-2461. doi: 10.1016/S0013-4686(01)00458-3
F. Croce, G.B. Appetecchi, L. Persi, B. Scrosati, Nature 394(1998) 456-458. doi: 10.1038/28818
X. Zhang, J. Xie, F. Shi, et al., Nano Lett. 18(2018) 3829-3838. doi: 10.1021/acs.nanolett.8b01111
H.J. Zhang, X.W. Zhang, E. Shiue, P.S. Fedkiw, J. Power Sources 177(2008) 561-565. doi: 10.1016/j.jpowsour.2007.11.064
W.W. Cui, D.Y. Tang, J. Appl. Polym. Sci. 126(2012) 510-518. doi: 10.1002/app.36804
Y.S. Lee, S.H. Ju, J.H. Kim, et al., Electrochem. Commun. 17(2012) 18-21. doi: 10.1016/j.elecom.2012.01.008
B.T. Zhao, G.C. Yuan, X. Chu, J.F. Yang, J. Zhao, Langmuir 34(2018) 6757-6765. doi: 10.1021/acs.langmuir.8b01195
E. Zygadlo-Monikowska, Z. Florjanczyk, E. Wielgus-Barry, J. Pasniewski, J. Power Sources 159(2006) 385-391. doi: 10.1016/j.jpowsour.2006.02.036
X.Y. Chen, D.P. Randall, C. Perruchot, et al., J. Colloid Interface Sci. 257(2003) 56-64. doi: 10.1016/S0021-9797(02)00014-0
J.C. Chai, Z.H. Liu, J. Ma, et al., Adv. Sci. 4(2017) 1600377. doi: 10.1002/advs.201600377
E. Quartarone, P. Mustarelli, A. Magistris, Solid State Ionics 110(1998) 1-14. doi: 10.1016/S0167-2738(98)00114-3
W.K. Shin, D.W. Kim, J. Power Sources 226(2013) 54-60. doi: 10.1016/j.jpowsour.2012.10.082
X.G. Sun, J. Hou, J.B. Kerr, Electrochim. Acta 50(2005) 1139-1147. doi: 10.1016/j.electacta.2004.08.011
G.S. MacGlashan, Y.G. Andreev, P.G. Bruce, Nature 398(1999) 792-794. doi: 10.1038/19730

Figure 1 TGA curves for the SiO2, initiator-functionalized silica particles, lithium polystyrene sulfonate brushes grafted silica particles.

Figure 2 Temperature dependent ionic conductivity of SiO2-LiPSS composite polymer electrolyte, SiO2 composite polymer electrolyte and PVC polymer electrolyte.

Figure 3 Current-time curve obtained from chronoamperometry at a DC polarization of 10 mV, inset: ESI response of Li/polymer electrolyte/Li cell before and after polarization.

Figure 4 Galvanostatic cycling curve of PVC, SiO2-PVC and SiO2-PVC-LiPSS composite polymer electrolyte at 0.10 mA/cm2.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:

 下载:
下载: