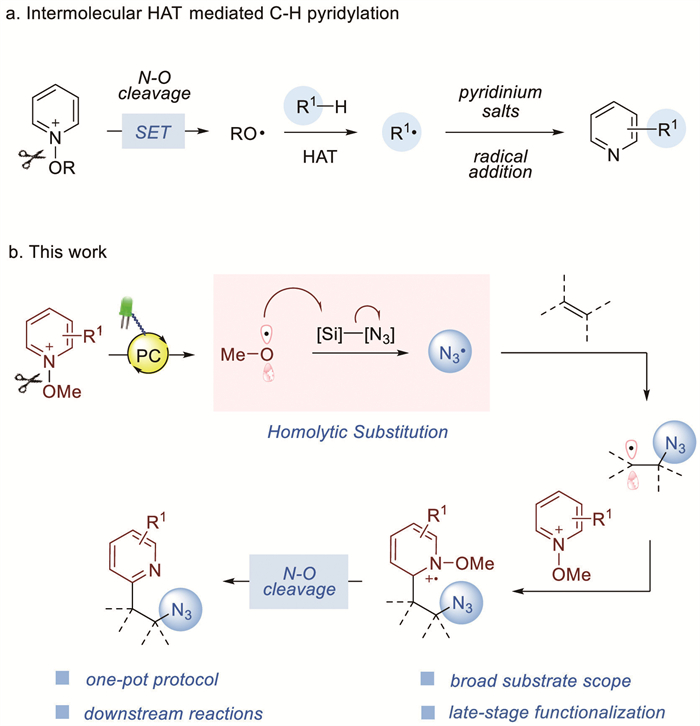
Scheme 1.
Methods for generating alkoxy radicals from N-substituted pyridinium salts and pyridylation reactions of these.
Visible light-mediated syntheses of β-pyridyl azides via three-component radical relay
Shan Wang , Ya-Jian Hu , Xuan Deng , Guang-Yi Zhang , Zichen Xu , Yu-Tao He

Pyridines are privileged cores frequently found in a wide-range of pharmaceuticals, functional materials, and natural products [1]. Accordingly, myriads of synthetic methods, such as encountered in Minisci-type reactions, have been developed to rapidly functionalize such scaffolds, and have provided ready access to the core structures of many medicinally important derivatives [2-4]. Despite their synthetic versatility, these methods demand stoichiometric amounts of Brønsted acids, and frequently resulted in mixtures of undesired byproducts due to competitive overalkylation [5,6]. Therefore, there is a growing demand for methods that can functionalize pyridine derivatives in a predictable and controllable manner. In this context, the advent of photocatalyzed reactions utilizing the bench-stable N-alkoxypyridinium salts as versatile pyridine surrogates marked a significant advance, which offers direct access to functionalized pyridine derivatives [7-10]. Relative to simple pyridines, the readily available N-alkoxypyridinium salts with low-lying LUMOs exhibit enhanced reactivity under acid-free conditions [11,12], and they have emerged as promising alkoxy radical precursors [13,14]. Indeed, efficient methods for functionalization by using N-alkoxypyridinium salts have been developed, and so demonstrating that the desired radical processes can be initiated by alkoxy radicals generated via light-promoted homolysis of the N–O bonds contained within N-alkoxypyridinium systems (Scheme 1a) [15-20]. For examples, Hong and co-workers reported various alkoxy radical-mediated intermolecular hydrogen atom transfer (HAT) strategies for the site-selective functionalization pyridines [21,22]. We recently disclosed a photochemical synthesis of unsymmetrical methylene-bridged bis-heterocycles involving an in situ generated O-centered radical-mediated HAT process using N-methoxypyridinium salts [23]. To broaden the scope of the synthetic strategy encapsulated by these pioneering studies, we considered whether methoxy radicals are capable of engaging in a radical relay process with trimethylsilyl azide (TMSN3) to generate azidyl radical, which could promote a multicomponent Minisci reaction.
Organic azides are used as highly powerful and valuable building blocks in synthetic organic chemistry because they can undergo a variety of chemical transformations [24], such as Staudinger reduction [25,26], “click” reactions [27], and others [28]. Traditional approaches for the generation and use of azidyl radical often depend on stoichiometric oxidant [29-33]. In this context, the development of an environmentally friendly and atom-economical process for regioselective azidation is greatly important [34,35]. Inspired by our recent interest in developing alkoxy radical-relay process promoted by visible light [36]. We speculated, as shown in Scheme 1b, that the reactive azidyl radical might be generated from TMSN3 via Si−N3 bond activation mediated by in situ generated methoxy radical. The alkyl radical intermediate was then formed as a result of the addition of azidyl radical to the alkene, which may set the stage for a second radical addition to the pyridine ring and so representing a potentially versatile extension of the classic Minisci reaction. To apply the strategy for such a reaction sequence, the alkyl radical intermediate should need to react preferentially with the pyridyl moiety of N-methoxypyridinium salts over radical−radical coupling with azidyl radical to generate the diazidation byproduct. In our working hypothesis, there is a large dynamic driving force for the formation of azidopyridylation prodcuts because of the relatively high concentration and strong electrophilic nature of N-methoxypyridinium salts [9,37]. Herein we detail the successful implementation of such a sequence. This method offers a facile access to valuable azidyl- and pyridyl-containing frameworks under mild reaction conditions.
To investigate the viability of the proposed radical relay reaction sequence, unactivated alkene 1a, N-methoxypyridinium salt 2a, and TMSN3 were selected as the model substrates under the irradiation of Green LEDs (λmax = 525 nm) to identify workable reaction conditions (see Supporting information for further details). After evaluating various photosensitizers, as shown in Table 1, we were pleased to find that the use of (Ir[dF(F)ppy]2(dCF3))PF6 as the photocatalyst in acetonitrile at room temperature afforded the β-pyridyl azide 4a in 48% yield (entry 6), thus indicating that the proposed overall process involving a radical azidation and subsequent pyridylation sequence can be achieved effectively. Among the solvents screened, acetonitrile was optimal since it showed higher efficiency (entries 6-8). The choice of base was critical for the reaction efficiency, and NaHCO3 was proved to be the optimum one by delivering 4a in 69% yield (entry 11). From the viewpoint of atom economy, oxidant free and operationally simple protocols are highly appealing. Thus, entry 11 was chosen as the optimal reaction conditions, even though the addition of oxidant resulted in a higher yield of 4a (entry 12). Interestingly, a low yield of product 4a (12%) was also obtained in the absence of the photocatalyst, indication that electron-donors might form EDA complexes with pyridinium salts and ultimately leading to the azidopyridylation of alkenes (entry 14) [38-41]. A control experiment confirmed that visible light is crucial (entry 15). Consistent with the proposed radical mechanism for the reaction, no desired product was observed when the reaction was conducted in the presence of TEMPO (entry 16).
 DownLoad:
CSV
DownLoad:
CSV
 |
||||
| Entry | Photocatalyst | Solvent | Base | Yield (%)b |
| 1 | 4CzIPN | MeCN | — | trace |
| 2 | Eosin Y | MeCN | — | 23 |
| 3 | Acr+ClO4- | MeCN | — | 18 |
| 4 | Ru(bpy)3Cl2·6H2O | MeCN | — | 21 |
| 5 | Ir(F-ppy)3 | MeCN | — | 25 |
| 6 | (Ir[dF(F)ppy]2(dCF3))PF6 | MeCN | — | 48 |
| 7 | (Ir[dF(F)ppy]2(dCF3))PF6 | 1,2-DCE | — | 41 |
| 8 | (Ir[dF(F)ppy]2(dCF3))PF6 | DMSO | — | 30 |
| 9 | (Ir[dF(F)ppy]2(dCF3))PF6 | MeCN | Et3N | 0 |
| 10 | (Ir[dF(F)ppy]2(dCF3))PF6 | MeCN | K3PO4 | 54 |
| 11 | (Ir[dF(F)ppy]2(dCF3))PF6 | MeCN | NaHCO3 | 69 |
| 12c | (Ir[dF(F)ppy]2(dCF3))PF6 | MeCN | NaHCO3 | 75 |
| 13c,d,e | — | MeCN | NaHCO3 | 20 |
| 14 | — | MeCN | NaHCO3 | 12 |
| 15e | (Ir[dF(F)ppy]2(dCF3))PF6 | MeCN | NaHCO3 | 0 |
| 16f | (Ir[dF(F)ppy]2(dCF3))PF6 | MeCN | NaHCO3 | 0 |
| a Reactions conditions: 1a (0.2 mmol), 2a (0.3 mmol), TMSN3 (0.4 mmol), NaHCO3 (0.3 mmol), photocatalyst (2.0 mol%), MeCN (1.0 mL), 40 W Kessil green LEDs (525 nm), 16 h. b Yield of isolated product. c (NH4)2S2O8 (1.5 equiv.) was added. d The reaction was carried out at 70 ℃. e The reaction was carried out in the dark. f TEMPO (2.0 equiv.) was added. |
||||
Once optimal reaction conditions had been established, we investigated the substrate scope to demonstrate the versatility of the current photocatalytic tandem reaction. Initially, a variety of alkene substrates were subjected to the optimized reaction conditions, as revealed in Scheme 2. Substrates bearing either electron-withdrawing or electron-donating groups on the aromatic ring proceeded evenly in the reaction with 2a and afforded the expected β-pyridyl azides (4a−4i). In addition, unactivated alkenes bearing esters (1j−1m), ethers (1n−1o), oxy-isoindolinedione (1p), phenyl (1q), alkyls (1r−1t), ketone (1u), amides (1v−1x), and trimethylsilane (1y) were well tolerated in this reaction. Later, the molecular structure of 4p was unambiguously confirmed by single-crystal X-ray analysis (see Supporting information for details). Gratifyingly, bromide was tolerable in this transformation to provide the azidopyridylation product 4z, thus allowing further modification. Notably, this methodology was found to be successful with enol ether (1aa) to yield the desired product. Moreover, to demonstrate the broad applicability of our developed method, the utility of the protocol in late-stage functionalisations was explored. For example, theobromine, fenofibric acid, (-)-menthol, 4-methylumbelliferone, a flavone-derivative, and (+)-sulbactam derivatives were all successfully engaged to afford the corresponding products (4ab-4ag), indicating the potential of this method for late-stage application.
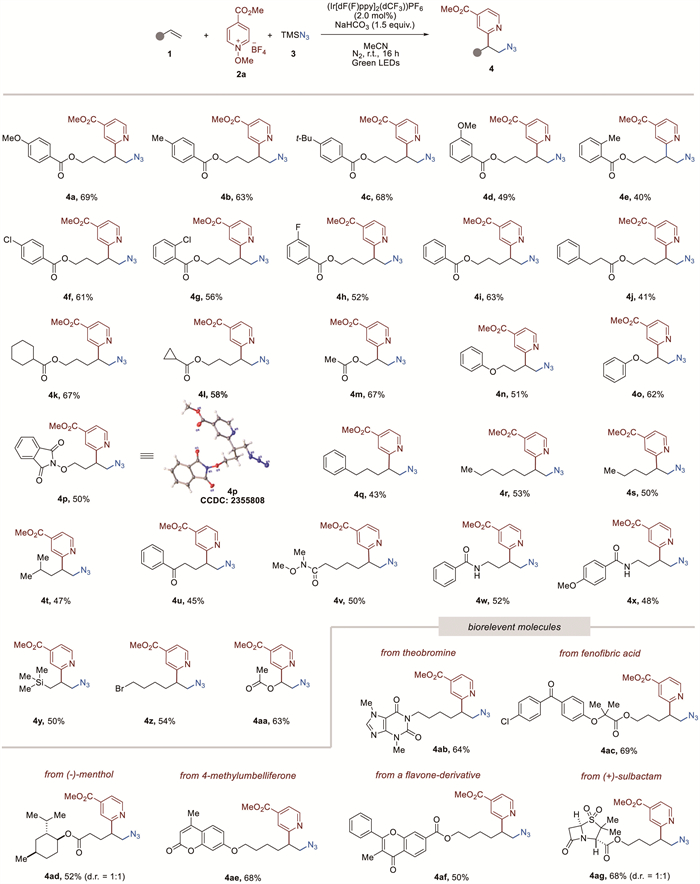
To further test the applicability of the radical relay process, a range of other heteroarenium salts to serve as coupling partners was explored, and the results are presented in Scheme 3. Reactions of pyridines bearing C-4 substituents (cyano, trifluoromethyl, and methyl ketone) proceeded smoothly to furnish the desired products (5a–5c). Pyridine bearing ester groups at the C-3 position deliver a 3:1 mixture compound 5d and its C-2 regioisomer in 71% combined yield. When substrates bearing electron-withdrawing groups on the pyridyl ring at the C-2 position (cyano, and trifluoromethyl) were subjected to the reaction, C-4 alkylation products 5e and 5f were exclusively obtained in 75% and 72% yields, respectively. Moreover, expanding the scope from the pyridines to the quinoline and isoquinoline systems were also adaptable. Specifically, quinolinium substituted with the cyano, methyl ketone, methoxy, fluoro, bromo, and phenyl at various positions were tolerated in this transformation, leading to the formation of the desired products (5h–5n). When isoquinolinium was used as the heteroarene source, the C-1 alkylation products 5o and 5p were formed, highlighting the broad functional group tolerance.
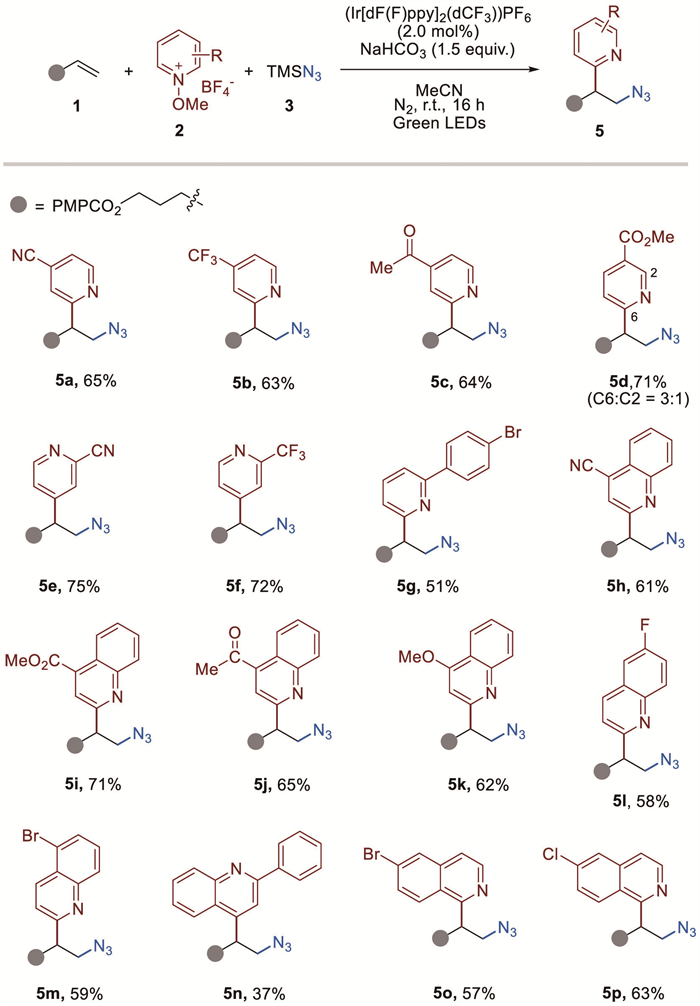
With this convenient approach to accessing β-pyridyl azides, further transformations of the product were surveyed to demonstrate the synthetic utility of this radical azidopyridylation protocol (Scheme 4). For instance, triazole 6 could be easily achieved from the derived β-pyridyl azide 4a and phenylacetylene through a copper catalyzed azide−alkyne [3 + 2] click reaction (Eq. 1) [42]. Treatment of 4a with acetylacetone in the presence of K2CO3 then delivering triazole product 7 in 82% yield (Eq. 2). Further, β-pyridyl azide 4a could be converted into β-pyridyl amine 8 via a phosphine-mediated Staudinger reduction (Eq. 3) [26].
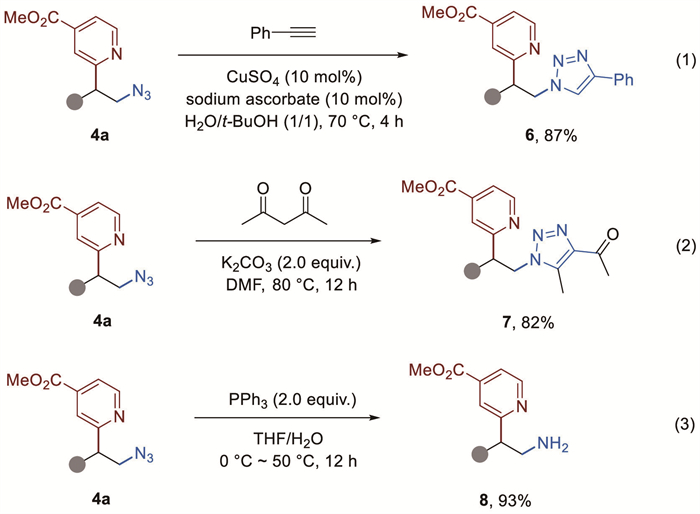
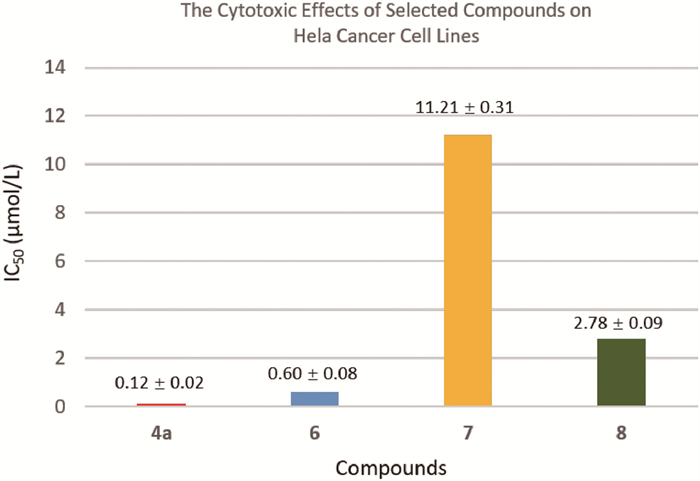
The ready access to the above-mentioned heterocycles offered by the protocols reported herein presented an opportunity to examine the cytotoxic effects of these selected compounds against HeLa, an immortalized cell line. As revealed in Fig. 1 (see Supporting information for further details), the compounds 4a, 6, 7 and 8 displayed IC50 values of 0.12 ± 0.02, 0.60 ± 0.08, 11.21 ± 0.31, and 2.78 ± 0.09 µmol/L, respectively. Therefore, these preliminary cell viability data suggest that these readily-derived compounds have potential as cancer therapeutic candidates.
To elucidate the reaction mechanism, a series of Stern−Volmer quenching experiments were performed [43,44], revealing that the quenching rate was directly proportional to the concentration of pyridinium salt 2a. On the other hand, the photoexcited Ir*catalyst was not quenched by 1a, TMSN3, and NaHCO3. The model reaction quantum yield was found to be
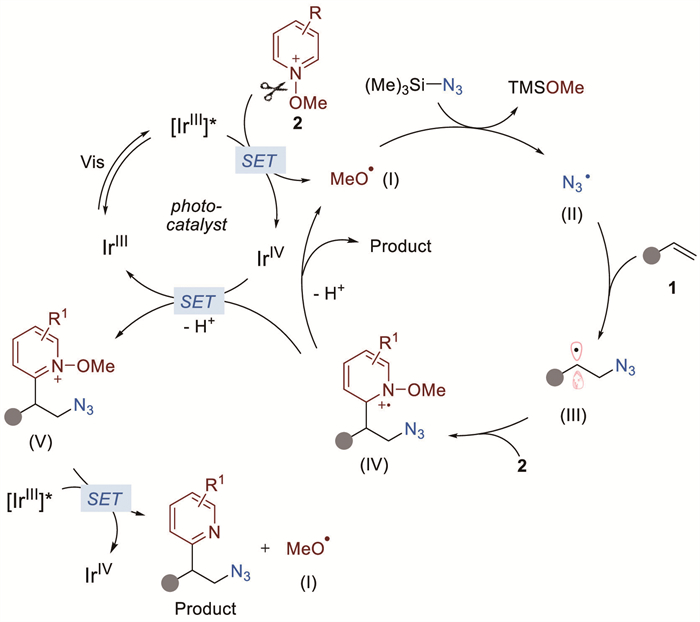
A possible mechanistic pathway is depicted in Scheme 5. Initially, the generated excited-state Ir* species facilitates the single-electron transfer (SET) reduction of pyridinium salt 2 to liberate the methoxy radical Ⅰ, which appears to react with TMSN3 to generate the reactive azidyl radical Ⅱ. Subsequently, the azidyl radical engages in an addition to the alkene 1 to provide radical intermediate Ⅲ. The resultant radical Ⅲ attacks pyridinium salt 2 in a Minisci-type process to form unstable radical cation Ⅳ. Finally, the radical cation Ⅳ undergoes deprotonation followed by homolytic cleavage of the N–O bond to furnish the observed β-pyridyl azide and methoxy radical Ⅰ. The released methoxy radical Ⅰ then promotes the radical-chain process with TMSN3, thus continuing the cycle.
In conclusion, we have presented a visible light-mediated three-component azidopyridylation of unactivated alkenes with accessible TMSN3 and pyridinium salts via an in situ generated methoxy radical-initiated relay reaction. This modular protocol features mild conditions, exhibits broad compatibility of functional groups and substrate scope, and highlights its potential in the late-stage diversification of biorelevent molecules. Employing this approach, a variety of synthetically useful azidyl- and pyridyl-containing building blocks were prepared. The resulting β-pyridyl azides could be efficiently converted into triazoles and β-pyridyl amine. This advancement is anticipated to have significant application potential in organic synthesis. Further mechanistic research and synthetic utilization of this chemistry are underway in our laboratories.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Shan Wang: Methodology, Investigation, Data curation. Ya-Jian Hu: Methodology, Investigation, Data curation. Xuan Deng: Data curation. Guang-Yi Zhang: Data curation. Zichen Xu: Writing – review & editing. Yu-Tao He: Writing – review & editing, Writing – original draft.
We thank Guangdong Basic and Applied Basic Research Foundation (Nos. 2022A1515110367, and 2024A1515010884), the National Natural Science Foundation of China (No. 22201104) and the Science and Technology Projects in Guangzhou (No. 2023A04J0079) for financial support.
Supplementary material associated with this article can be found, in the online version, at doi:
P. Bhutani, G. Joshi, N. Raja, et al., J. Med. Chem. 64 (2021) 2339–2381. doi: 10.1021/acs.jmedchem.0c01786
G.X. Li, C.A. Morales-Rivera, Y. Wang, et al., Chem. Sci. 7 (2016) 6407–6412. doi: 10.1039/C6SC02653B
R.S. Proctor, R.J. Phipps, Angew. Chem. Int. Ed. 58 (2019) 13666–13699. doi: 10.1002/anie.201900977
J.F. Yang, Y.F. Liu, L.L. Wei, et al., J. Org. Chem. 89 (2024) 4249–4260. doi: 10.1021/acs.joc.4c00093
S. Caron, R.W. Dugger, S.G. Ruggeri, J.A. Ragan, D.H.B. Ripin, Chem. Rev. 106 (2006) 2943–2989. doi: 10.1021/cr040679f
R.S. Proctor, P. Chuentragool, A.C. Colgan, R.J. Phipps, J. Am. Chem. Soc. 143 (2021) 4928–4934. doi: 10.1021/jacs.1c01556
I. Kim, B. Park, G. Kang, et al., Angew. Chem. Int. Ed. 57 (2018) 15517–15522. doi: 10.1002/anie.201809879
F.S. He, S. Ye, J. Wu, ACS Catal. 9 (2019) 8943–8960. doi: 10.1021/acscatal.9b03084
M. Kim, Y. Koo, S. Hong, Acc. Chem. Res. 55 (2022) 3043–3056. doi: 10.1021/acs.accounts.2c00530
Y. Wang, Y. Bao, M. Tang, et al., Chem. Commun. 58 (2022) 3847–3864. doi: 10.1039/d2cc00369d
X. Ma, H. Dang, J.A. Rose, P. Rablen, S.B. Herzon, J. Am. Chem. Soc. 139 (2017) 5998–6007. doi: 10.1021/jacs.7b02388
Y.T. He, D. Kang, I. Kim, S. Hong, Green Chem. 20 (2018) 5209-5214.
A.L. Barthelemy, B. Tuccio, E. Magnier, G. Dagousset, Angew. Chem. Int. Ed. 57 (2018) 13790–13794. doi: 10.1002/anie.201806522
F. Rammal, D. Gao, S. Boujnah, et al., Org. Lett. 22 (2020) 7671–7675. doi: 10.1021/acs.orglett.0c02863
V. Quint, F. Morlet-Savary, J.F. Lohier, et al., J. Am. Chem. Soc. 138 (2016) 7436–7441. doi: 10.1021/jacs.6b04069
Y. Kim, K. Lee, G. R. Mathi, I. Kim, S. Hong, Green Chem. 21 (2019) 2082-2087.
V. Quint, N. Chouchène, M. Askri, et al., Org. Chem. Front. 6 (2019) 41–44. doi: 10.1039/c8qo00985f
A. Inial, F. Morlet-Savary, J. Lalevée, A.C. Gaumont, S. Lakhdar, Org. Lett. 22 (2020) 4404–4407. doi: 10.1021/acs.orglett.0c01409
R. Laha, T.I. Patel, M.J. Moschitto, Org. Lett. 24 (2022) 7394–7399. doi: 10.1021/acs.orglett.2c02932
D. Xie, Y. Wang, X. Zhang, Z. Fu, D. Niu, Angew. Chem. Int. Ed. 61 (2022) e202204922. doi: 10.1002/anie.202204922
S. Jung, H. Lee, Y. Moon, H.Y. Jung, S. Hong, ACS Catal. 9 (2019) 9891–9896. doi: 10.1021/acscatal.9b03367
I. Kim, G. Kang, K. Lee, et al., J. Am. Chem. Soc. 141 (2019) 9239–9248. doi: 10.1021/jacs.9b02013
T.T. Yuan, J. Chen, S. Paul, et al., Org. Chem. Front. 10 (2023) 4649–4657. doi: 10.1039/d3qo00754e
S. Bräse, K. Banert, Organic azides, Synthesis and Applications, Wiley, New York, 2010.
H.A. van Kalkeren, J.J. Bruins, F.P. Rutjes, F.L. van Delft, Adv. Synth. Catal. 354 (2012) 1417–1421. doi: 10.1002/adsc.201100967
X. Li, Z. Wang, W. Luo, et al., Molecules 27 (2022) 5707–5717. doi: 10.3390/molecules27175707
P. Thirumurugan, D. Matosiuk, K. Jozwiak, Chem. Rev. 113 (2013) 4905–4979. doi: 10.1021/cr200409f
S. Bräse, C. Gil, K. Knepper, V. Zimmermann, Angew. Chem. Int. Ed. 44 (2005) 5188–5240. doi: 10.1002/anie.200400657
K. Wu, Y. Liang, N. Jiao, Molecules 21 (2016) 352–372. doi: 10.3390/molecules21030352
Z. Liu, Z.Q. Liu, Org. Lett. 19 (2017) 5649–5652. doi: 10.1021/acs.orglett.7b02788
P. Sivaguru, Y. Ning, X. Bi, Chem. Rev. 121 (2021) 4253–4307. doi: 10.1021/acs.chemrev.0c01124
Y. Luo, L. Lv, Z. Li, Org. Lett. 24 (2022) 8052–8056. doi: 10.1021/acs.orglett.2c03302
E.R. Lopat’eva, I.B. Krylov, S.A. Paveliev, et al., J. Org. Chem. 88 (2023) 13225–13235. doi: 10.1021/acs.joc.3c01470
J. Chen, S. Zhu, J. Qin, D.L. Chu, Chem. Commun. 55 (2019) 2336–2339. doi: 10.1039/c9cc00241c
C.X. Xia, Z. Li, R. Ye, et al., Org. Lett. 26 (2024) 3530–3535. doi: 10.1021/acs.orglett.4c00911
Y.J. Hu, J.C. Liu, G.Y. Zhang, et al., Org. Chem. Front. 11 (2024) 5016–5025. doi: 10.1039/d4qo00888j
J. Liu, H.W. Jiang, X.Q. Hu, P.F. Xu, Org. Lett. 26 (2024) 3661–3666. doi: 10.1021/acs.orglett.4c01186
S. Jung, S. Shin, S. Park, S. Hong, J. Am. Chem. Soc. 142 (2020) 11370–11375. doi: 10.1021/jacs.0c04499
M. Kim, E. You, S. Park, S. Hong, Chem. Sci. 12 (2021) 6629–6637. doi: 10.1039/d1sc00776a
S. Shin, S. Lee, W. Choi, N. Kim, S. Hong, Angew. Chem. Int. Ed. 60 (2021) 7873–7879. doi: 10.1002/anie.202016156
S. Hong, B. Kweon, W. Lee, S. Chang, S. Hong, Org. Lett. 25 (2023) 2722–2727. doi: 10.1021/acs.orglett.3c00922
Y.T. Shen, Y.S. Ran, B. Jiang, et al., Org. Lett. 25 (2023) 4525–4529. doi: 10.1021/acs.orglett.3c01562
M.A. Cismesia, T.P. Yoon, Chem. Sci. 6 (2015) 5426–5434. doi: 10.1039/C5SC02185E
D.M. Fischer, H. Lindner, W.M. Amberg, E.M. Carreira, J. Am. Chem. Soc. 145 (2023) 774–780. doi: 10.1021/jacs.2c11680

Scheme 1 Methods for generating alkoxy radicals from N-substituted pyridinium salts and pyridylation reactions of these.

Scheme 2 Substrate scope of unactivated alkenes. Reaction conditions: 1 (0.2 mmol), 2a (0.3 mmol), 3 (0.4 mmol), NaHCO3 (0.3 mmol), (Ir[dF(F)ppy]2(dCF3))PF6 (2.0 mol%), MeCN (1.0 mL). 40 W Kessil green LEDs (525 nm), 16 h. Isolated yields.

Scheme 3 Substrate scope of heteroarenium salts. Reaction conditions: 1a (0.2 mmol), 2 (0.3 mmol), 3 (0.4 mmol), NaHCO3 (0.3 mmol), (Ir[dF(F)ppy]2(dCF3))PF6 (2.0 mol%), MeCN (1.0 mL). 40 W Kessil green LEDs (525 nm), 16 h. Isolated yields.
Table 1. Optimization of the reaction conditions.a
|
||||
| Entry | Photocatalyst | Solvent | Base | Yield (%)b |
| 1 | 4CzIPN | MeCN | — | trace |
| 2 | Eosin Y | MeCN | — | 23 |
| 3 | Acr+ClO4- | MeCN | — | 18 |
| 4 | Ru(bpy)3Cl2·6H2O | MeCN | — | 21 |
| 5 | Ir(F-ppy)3 | MeCN | — | 25 |
| 6 | (Ir[dF(F)ppy]2(dCF3))PF6 | MeCN | — | 48 |
| 7 | (Ir[dF(F)ppy]2(dCF3))PF6 | 1,2-DCE | — | 41 |
| 8 | (Ir[dF(F)ppy]2(dCF3))PF6 | DMSO | — | 30 |
| 9 | (Ir[dF(F)ppy]2(dCF3))PF6 | MeCN | Et3N | 0 |
| 10 | (Ir[dF(F)ppy]2(dCF3))PF6 | MeCN | K3PO4 | 54 |
| 11 | (Ir[dF(F)ppy]2(dCF3))PF6 | MeCN | NaHCO3 | 69 |
| 12c | (Ir[dF(F)ppy]2(dCF3))PF6 | MeCN | NaHCO3 | 75 |
| 13c,d,e | — | MeCN | NaHCO3 | 20 |
| 14 | — | MeCN | NaHCO3 | 12 |
| 15e | (Ir[dF(F)ppy]2(dCF3))PF6 | MeCN | NaHCO3 | 0 |
| 16f | (Ir[dF(F)ppy]2(dCF3))PF6 | MeCN | NaHCO3 | 0 |
| a Reactions conditions: 1a (0.2 mmol), 2a (0.3 mmol), TMSN3 (0.4 mmol), NaHCO3 (0.3 mmol), photocatalyst (2.0 mol%), MeCN (1.0 mL), 40 W Kessil green LEDs (525 nm), 16 h. b Yield of isolated product. c (NH4)2S2O8 (1.5 equiv.) was added. d The reaction was carried out at 70 ℃. e The reaction was carried out in the dark. f TEMPO (2.0 equiv.) was added. |
||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们



 下载:
下载: