Joint Laboratory for Translational Cancer Research of Chinese Medicine of the Ministry of Education of the People’s Republic of China, International Institute for Translational Chinese Medicine, Guangzhou University of Chinese Medicine, Guangzhou 510006, China
b.
Guangzhou Yuemei Pharmaceutical Technology Co., Ltd., Guangzhou 510535, China
c.
Affiliated Foshan Maternity & Child Healthcare Hospital, Southern Medical University, Foshan 528000, China
d.
School of Pharmaceutical Sciences, Southern Medical University, Guangzhou 510515, China
e.
Department of Laboratory Medicine, Dongguan Institute of Clinical Cancer Research, Dongguan Key Laboratory of Precision Diagnosis and Treatment for Tumors, The Tenth Affiliated Hospital of Southern Medical University (Dongguan People’s Hospital), Dongguan 523058, China
* Corresponding authors. E-mail addresses: shtwang@ipe.ac.cn (S. Wang)
Received Date:
16 November 2023 Accepted Date:
08 January 2024 Revised Date:
04 January 2024 Available Online:
15 October 2024
Abstract:
Hepatocellular carcinoma is a common and fatal malignancy for which there is no effective systemic therapeutic strategy. Dihydroartemisinin (DHA), a derivative of artemisinin, has been shown to exert anti-tumor effects through the production of reactive oxygen species (ROS) and resultant mitochondrial damage. However, clinical translation is limited by several drawbacks, such as insolubility, instability and low bioavailability. Here, based on a nanomedicine-based delivery strategy, we fabricated mitochondria-targeted carrier-free nanoparticles coupling DHA and triphenylphosphonium (TPP), aiming to improve bioavailability and mitochondrial targeting. DHA-TPP nanoparticles can be passively delivered to the tumor site by enhanced penetration and retention and then internalized. Flow cytometry and Western blot analysis showed that DHA-TPP nanoparticles increased intracellular ROS, which increased mitochondrial stress and in turn upregulated the downstream Bcl-2 pathway, leading to apoptosis. In vivo experiments showed that DHA-TPP nanoparticles exhibited anti-tumor effects in a mouse model of hepatocellular carcinoma. These findings suggest carrier-free DHA-TPP nanoparticles as a potential therapeutic strategy for hepatocellular carcinoma.
Hepatocellular carcinoma (HCC) is a frequent liver malignancy and causes 830,000 deaths per year, making it the third leading cause of cancer death [1,2]. Surgical treatment can improve survival rates; however, the risk of liver decompensation and recurrence reduces patients’ quality of life. Moreover, for patients with advanced-stage HCC, systemic therapy is not ideal because of a lack of effective drug treatment. Therefore, there is an urgent need to develop novel therapies for HCC.
As a strategy for the discovery of anti-tumor drugs, the new use of old medicine has gained increasing attention [3]. The advantage of this process is that it reduces the cost and time to market drugs compared to traditional novel drug development [4]. Natural products, particularly in traditional Chinese medicine (TCM), have always been an important direct and indirect source of anti-tumor drugs [5,6]. Dihydroartemisinin (DHA), a first-line antimalarial drug, also shows great potential in the field of tumor therapy [7-10]. DHA is thought to generate reactive oxygen species (ROS) by scavenging peroxide bridges and promoting Fenton’s reaction [11]. Excessive ROS and mitochondrial stress interact with positive feedback to activate the Bcl-2 pathway and trigger apoptosis [12,13]. However, low water solubility and poor bioavailability limited the clinical application of DHA as an anti-cancer agent. To overcome these drawbacks and enhance clinical translational efficiency, the use of DHA-encapsulated nano-formulations has been proposed. In particular, the conversion of DHA into nanomedicine seems to be a promising strategy to improve stability and solubility, since nanoplatforms are prone to accumulate in tumor sites due to the enhanced permeability and retention (EPR) effect [14-16].
Despite their promise, nanoplatforms face significant challenges: complex preparation processes, insufficient loading efficiency and uncontrolled drug release [17]. Moreover, another concern is whether the degradation products of nanocarriers lead to adverse effects [18,19]. Fortunately, emerging carrier-free delivery systems can be prepared through a simple self-assembly process without the use of nanocarriers, avoiding the above shortcomings [20,21].
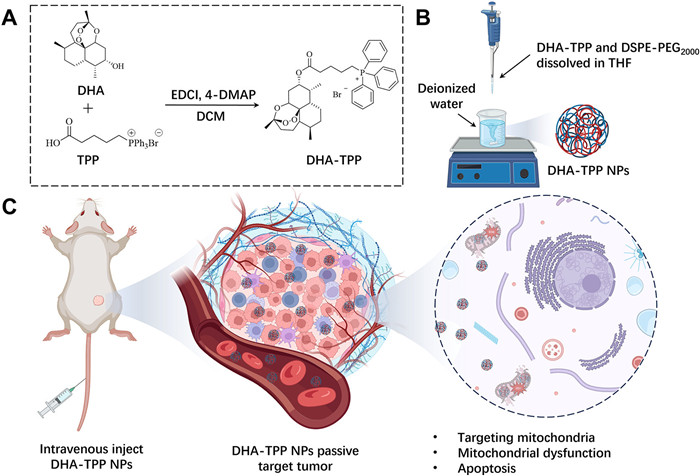
To the best of our knowledge, the self-assembled DHA-based nanoparticles for cancer therapy that have been reported all fail to deliver precisely to the target [22-24]. Mitochondria play a crucial role in the production of cellular energy. Hence, the development of mitochondria-targeted nano-drug delivery systems holds great potential for tumor therapy [25-27]. In this study, to enhance the anti-tumor efficacy of DHA, carrier-free DHA-triphenylphosphonium (TPP) nanoparticles (DHA-TPP NPs, Fig. 1) were designed by the conjugation of DHA and TPP, a mitochondrial-targeting ligand [28-30].
Figure 1
Figure 1.
Preparation of DHA-TPP NPs and proposed anti-tumor mechanism. The fabricated NPs can be accumulated in the tumor site through the EPR effect and internalized by tumor cells. Subsequently, DHA-TPP NPs are targeting mitochondria, thereby inducing mitochondrial dysfunction and activating the apoptosis pathway.
DHA-TPP was synthesized via esterification from DHA and TPP in a 75% yield (details see Fig. S1 in Supporting information). The structure of the target compound was confirmed by nuclear magnetic reasonance (NMR) and mass spectra (Figs. S2–S4 in Supporting information). The characteristic peaks at δ 7.67–7.90 ppm in 1H NMR and the characteristic peaks at 171.70, 135.00, 133.78, 130.51 and 118.15 ppm in 13C NMR spectra confirmed the successful introduction of TPP. In addition, the molecular weight of 629.37 (M-Br)+ (Fig. S4) in MS spectra demonstrated the correctness of DHA-TPP again.
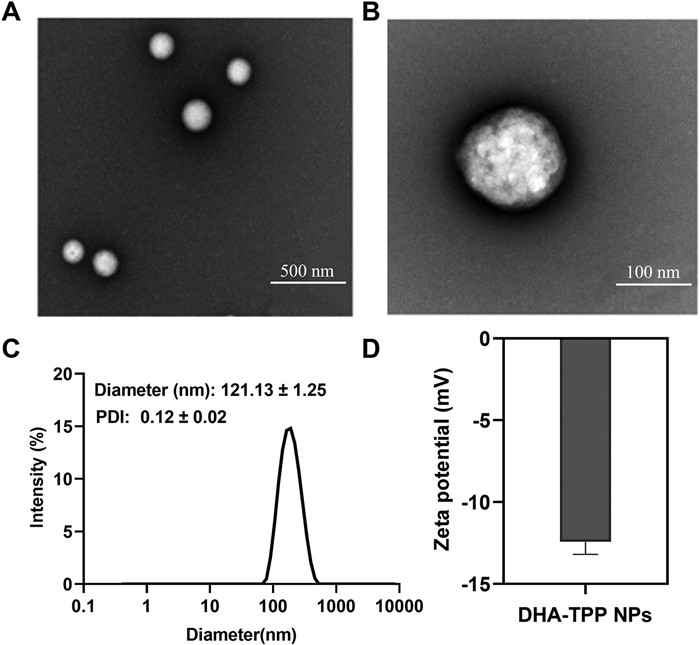
DHA-TPP NPs were prepared by nanoprecipitation. DHA-TPP is an amphiphilic molecule that spontaneously aggregates in aqueous environments without the use of exogenous excipients. Polyethylene glycol molecules were added to increase the stability of nanomedicine and extend blood circulation time. As shown in Fig. S5 (Supporting information), the DHA-TPP NPs exhibit outstanding drug loading capability, demonstrating the advantage of carrier-free nanomedicine. The morphologies of DHA-TPP NPs were examined by transmission electron microscope (TEM). As shown in Figs. 2A and B, uniformly sphere was observed. The average particle size was about 121.13 nm and the zeta-potential was −11 mV (Figs. 2C and D). Stability is a key factor in the clinical application of nanomedicines to assure efficacy and safety [31]. During a 7-day stability test, polymer dispersity index (PDI) and particle size of DHA-TPP NPs increased slightly, but not significantly (Fig. S6 in Supporting information). The PDI of nanoparticles in 10% fetal bovine serum (FBS) culture medium is slightly higher than that in pure water, which may cause the formation of a DHA-TPP NPs corona complex in FBS. To prevent the binding of proteins and NPs in serum, 1,2-distearoyl-sn–glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)−2000] (DSPE-PEG2000) was added to coat DHA-TPP NPs with a polymer film and improve their stability [32]. These results indicate that DHA-TPP monomers can form stable DHA-TPP NPs through self-assembly.
Figure 2
Figure 2.
Characterization of DHA-TPP NPs. (A, B) Representative TEM images of DHA-TPP NPs. Scale bars: 500 nm (A) and 100 nm (B). (C) Particle size of DHA-TPP NPs. (D) Zeta-potential of DHA-TPP NPs. Data are expressed as mean ± standard deviation (SD) (n = 3).
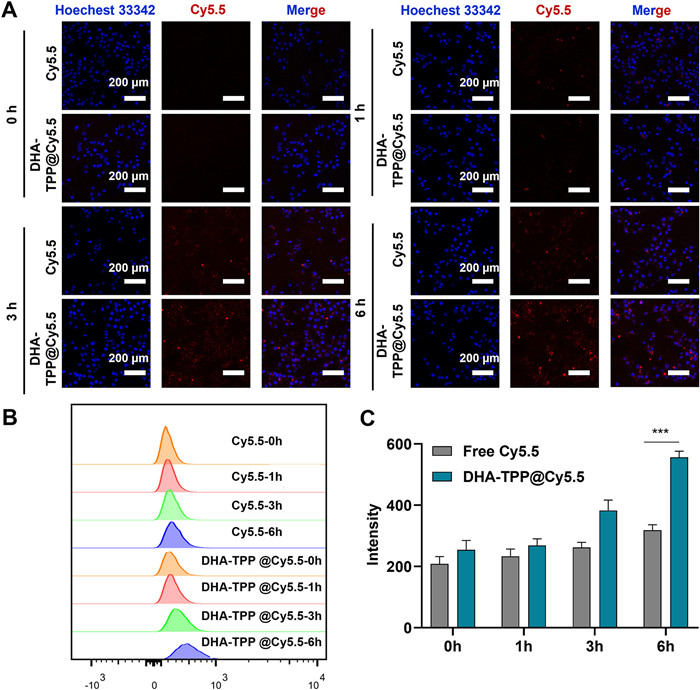
Internalization of DHA-TPP NPs in HepG2 cells was examined by flow cytometry and confocal laser scanning microscopy (CLSM). In this experiment, Cy5.5 was used to label DHA-TPP NPs and Hoechst 33342 was used to stain nuclei. As shown in Figs. 3A and B, intracellular red fluorescence was enhanced in a time-dependent manner. The intracellular red fluorescent of DHA-TPP@Cy5.5 NPs group was stronger than the free Cy5.5 group at 3 and 6 h, indicating that NPs are more easily internalized by HepG2 cells.
Figure 3
Figure 3.
Cellular uptake analysis. (A) Fluorescent images after incubating HepG2 cells with Cy5.5, DHA-TPP@Cy5.5 for 0, 1, 3, 6 h. (B, C) Flow cytometry analysis of cellular uptake capability. Data are expressed as mean ± SD (n = 3), ***P < 0.001.
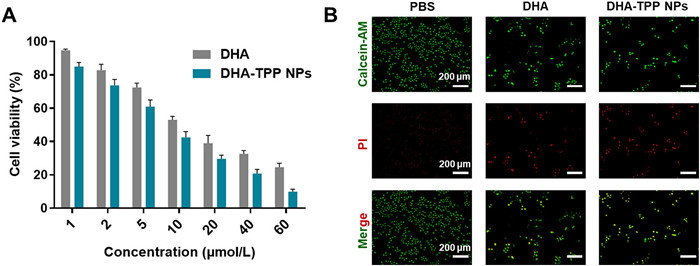
Subsequently, the anti-tumor activities of free DHA and DHA-TPP NPs were examined using MTT assay. After 48 h of incubation, both DHA-TPP NPs and free DHA could effectively inhibit the viability of HepG2. In DHA-TPP NPs group, over 90.15% of HepG2 cells viability was inhibited at a dose of 60 µmol/L, while free DHA reduced viability by 75.48% at the same concentration (Fig. 4A). Next, the anti-tumor efficacy of free DHA and DHA-TPP NPs were further determined using a LIVE/DEAD assay. As depicted in Fig. 4B, green fluorescence indicates living cells and red fluorescence indicates dead cells. The green fluorescence in the DHA-TPP NPs sample is weaker than that of the free DHA group, while the red fluorescence intensity in DHA-TPP NPs is stronger than that in the free DHA. Collectively, DHA-TPP NPs exhibited more effective anti-tumor activity against HepG2 cells, compared with the DHA.
Figure 4
Figure 4.
Anti-tumor activity against HepG2 cells. (A) HepG2 viability after incubation with various concentrations of DHA and DHA-TPP NPs for 48 h. (B) Calcein-AM/PI staining of HepG2 cells after treatment with DHA and DHA-TPP NPs for 24 h, respectively. Data are expressed as mean ± SD (n = 3).
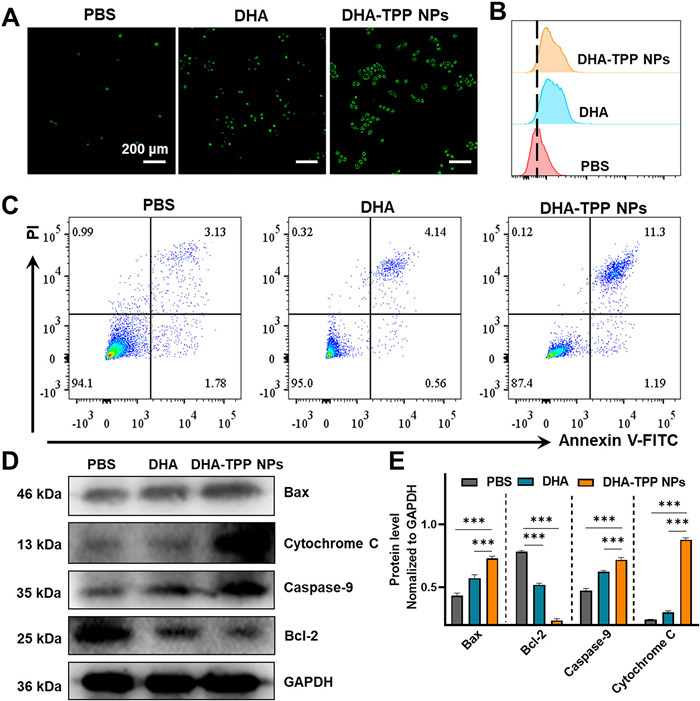
Increasing evidence indicates that the anti-tumor mechanism of artemisinin and its derivatives involves excessive ROS production; thus, we measured intracellular ROS in HepG2 cells that were treated with DHA and DHA-TPP NPs. As shown in Figs. 5A and B, green fluorescence, which indicates ROS level, in DHA-treated cells was significantly stronger than that of the phosphate buffer saline (PBS) group, and remarkably weaker than that of DHA-TPP NPs treated cells. Excessive ROS is an inducer of mitochondrial stress, which leads to mitochondrial damage, resulting in the release of cytochrome C, accompanied by a down-regulation of Bcl-2 and an up-regulation of Bax. Finally, caspases are activated, promoting apoptotic body formation and triggering apoptosis. Therefore, we next investigated the effect of DHA-TPP NPs on apoptosis. Flow cytometry with Annexin V/propidium iodide (PI) staining was used to quantify early and late apoptotic cells by detecting changes in morphology. As shown in Fig. 5C, DHA (40 µmol/L) induced 4.70% apoptosis rate, while DHA-TPP NPs (40 µmol/L) induced 12.49% apoptosis, suggesting that DHA-TPP NPs are more potent than DHA in the induction of cell death. Western blot analysis results showed that DHA-TPP are better able to upregulate Bax and downregulate Bcl-2, thus decreasing the ratio of Bcl-2/Bax, resulting in cytochrome C release and apoptosis (Fig. 5D). Together, these results indicate that the anti-tumor mechanism of DHA is inducing apoptosis through production of excessive ROS, and show that carrier-free DHA-TPP NPs have a higher potency due to its mitochondria-targeting capability.
Figure 5
Figure 5.
Anti-tumor mechanism analysis. (A) Determination of intracellular ROS contents in HepG2 cells with different treatments at 12 h (Scale bar: 100 µm). (B) Flow cytometric analysis of ROS generation in HepG2 cells with different treatments at 12 h. (C) Annexin V-FITC apoptosis detection in different samples. (D) Western blot analysis of Bax, cytochrome C, caspase-9 and Bcl-2 in HepG2 cells after treatment with 20 µmol/L DHA and DHA-TPP NPs for 24 h. (E) semi-quantitative analysis of Bax, cytochrome C, caspase-9 and Bcl-2 expression levels. Data are expressed as mean ± SD (n = 3), ***P < 0.001. GAPDH, recombinant glyceraldehyde-3-phosphate dehydrogenase.
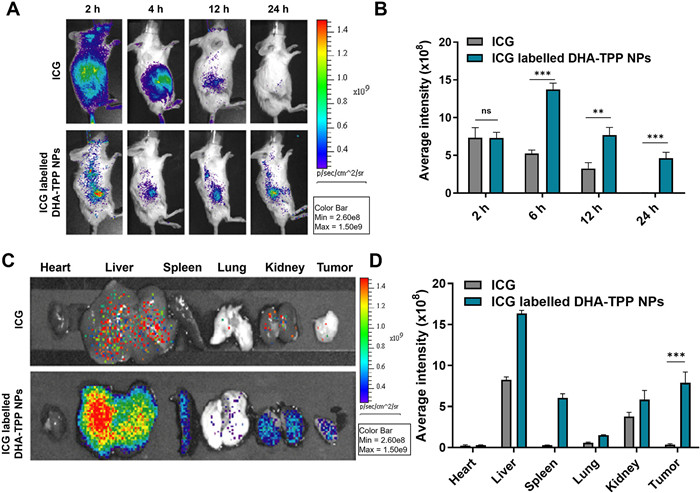
Due to the anti-tumor effects of NPs being associated with the degree of accumulation in lesions, we investigated the biodistribution of DHA-TPP NPs in tumor-bearing mice. The animal protocol for this study was approved by the Guangzhou University of Traditional Chinese Medicine Animal Care and Use Committee, and all of these mice were maintained under specific pathogen-free conditions (License number: SYXK (Guangzhou) 2019-0144). Free indocyanine green angiography (ICG) and ICG-labelled DHA-TPP NPs were intravenously injected, and in vivo fluorescence imaging was used at different 2, 4, 12 and 24 h, respectively. As shown in Figs. 6A and B, fluorescence in the free ICG group was seen throughout the entire body at 2 h, followed by a rapid decline. In contrast, the ICG-labelled DHA-TPP NPs group had a slower fluorescence decline and a higher efficiency of tumor accumulation over 24 h. The biodistributions of free ICG and ICG-labelled DHA-TPP NPs in major organs and tumors were then assessed. As shown in Figs. 6C and D, fluorescence intensities of heart, liver, spleen, lung, kidney and tumor in the ICG-labelled DHA-TPP NPs group were respectively 1.28, 1.98, 22.05, 2.55, 1.55, 22.9 times those of the free ICG group. This indicates that, compared to free drugs, DHA-TPP NPs can better aggregate at tumor sites due to the EPR effect and long circulation.
Figure 6
Figure 6.
Biodistribution of DHA-TPP NPs. (A) Representative in vivo fluorescence images of tumor-bearing mice at 2, 6, 12 and 24 h using ICG, ICG-labelled DHA-TPP NPs. (B) Semi-quantitative analysis of the fluorescence intensity in a tumor at designated time points after indicated treatments. (C) Ex vivo fluorescence images and (D) semi-quantitative of major organs from H22 tumor-bearing mice at 24 h. Data are expressed as mean ± SD (n = 3). **P < 0.01, ***P < 0.001.
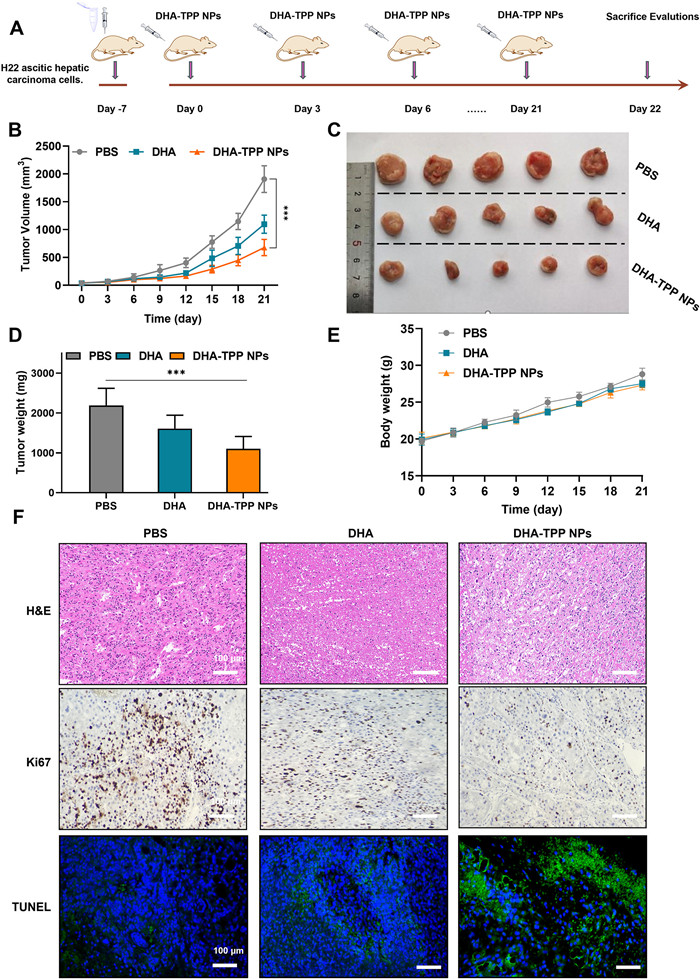
To verify the feasibility of intravenous injection of DHA and DHA-TPP NPs, we first performed a hemolysis assay. As shown in Fig. S7 (Supporting information), no hemolysis occurred in our preparations, illustrating that NPs were suitable for intravenous injection. The in vivo anti-tumor effects of free DHA and DHA-TPP NPs were then examined in a tumor-bearing mice model, established through subcutaneous injection of H22 cells. Tumor-bearing mice were randomly divided into three groups after tumor volume reached ~100 mm3, and then treated with PBS, free DHA or DHA-TPP NPs (Fig. 7A). Tumor volumes of all mice were recorded every three days. Tumor growth curves (Fig. 7B) show that tumor growth was moderately suppressed in the free DHA group compared with the PBS group due to the inherent toxicity of DHA, while this inhibition was more significant in the DHA-TPP NPs group indicating enhanced anti-tumor effect compared to free DHA. A similar phenomenon can also be observed when comparing average tumor weights from different groups. For the DHA and DHA-TPP NPs groups, average tumor weights were 0.73 and 0.50 times of the PBS group, respectively (Figs. 7C and D) while body weight showed no significant difference (Fig. 7E). Next, we evaluated the proliferation and apoptosis of tumor cells via histochemistry (Fig. 7F). Compared to the other two groups, significant apoptosis of tumor cells and weaker proliferative potential were observed in the DHA-TPP NPs group. This outcome can be interpreted from a variety of perspectives. First, the anti-tumor efficacy of DHA-TPP NPs was greatly enhanced compared to free DHA, which has been demonstrated in vitro. Second, DHA-TPP NPs exhibit both an EPR effect and stability in circulation and thus accumulate at the tumor site. Third, DHA-TPP NPs have outstanding solubility and are convenient for one-time large dose administration.
Figure 7
Figure 7.
Anti-tumor analysis in vivo. (A) Schematic illustration of in vivo pharmacodynamic study. (B) Tumor volume growth curves for different groups of mice after various treatments. (C) Photographs of excised tumors. (D) Average ex vivo tumor weights. (E) Body weight changes in tumor-bearing mice. (F) H&E, Ki67 and terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL) staining of tumor tissues after various treatments. Data are expressed as mean ± SD (n = 5), ***P < 0.001.
The biocompatibility of DHA-TPP NPs was then assessed through biochemical indexes and hematoxylin and eosin (H&E) staining of main organs (Fig. S8 in Supporting information). None of the test groups showed a significant increase in inflammatory cells or tissue damage, indicating that at the test dose used, DHA-TPP NPs do not cause any apparent histological abnormalities. Finally, we measured liver function parameters including alanine aminotransferase (ALT, IU/L), aspartate aminotransferase (AST, IU/L) and kidney index including blood urea nitrogen (BUN, mmol/L), and creatine (CREA, mmol/L), as well as heart function index including creatine kinase (Ck, IU/L). Among the three test groups, there was no significant difference in the levels of ALT, AST, BUN, CREA or Ck. Thus, DHA-TPP NPs are unlikely to cause general toxicity in mice at the test dose used, indicating they can be effectively and safely used for in vivo anti-tumor therapy.
To conclude, we have successfully synthesized DHA-TTP, which is a derivative of DHA that has a mitochondrial targeting function. We then converted this compound into carrier-free NPs utilizing a self-assembly method. In vitro experiments showed that these DHA-TPP NPs had satisfactory anti-tumor efficacy due to the promotion of mitochondrial dysfunction and activation of apoptosis. In vivo experiments indicated that DHA-TPP NPs can passively target the tumor site, thereby exerting anti-tumor efficacy. It is important to note that there are no significant toxic effects at therapeutic doses. To summarize, the prepared DHA-TPP NPs have several key advantages, including uniform morphology, high stability, effective drug loading, satisfactory anti-tumor efficacy and strong biocompatibility. Therefore, this study presents a cost-effective and efficient approach for the conversion of DHA into a carrier-free, mitochondria-targeting nanomedicine for the treatment of HCC.
Declaration of competing interest
The authors declare that they have no competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This study was funded and supported by the Department of Science and Technology of Guangdong Province (No. 2022B1111020005), Key Laboratory of Guangdong Provincial Food and Drug Administration (No. 2021ZDB03), the 2020 Guangdong Provincial Science and Technology Innovation Strategy Special Fund (Guangdong-Hong Kong-Macau Joint Lab, No. 2020B1212030006), Guangdong Province Universities and Colleges Pearl River Scholar Funded Scheme (No. Guochao Liao; 2019) and Guangdong Basic and Applied Basic Research Foundation (Nos. 2020B1515130005, 2022A1515110270, 202201011563), The Bureau of Science and Technology of Guangzhou City (No. HMJH2019000).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2024.109519.
C. Xu, L. Xiao, X. Zhang, et al., Bioorg. Med. Chem. Lett. 39 (2021) 127912. doi: 10.1016/j.bmcl.2021.127912
[31]
S. Đorđević, M.M. Gonzalez, I. ConejosSánchez, et al., Drug Deliv. Transl. Res. 12 (2022) 500–525. doi: 10.1007/s13346-021-01024-2
[32]
M. Marwa, S.A.L. Amr, S. Taro, et al., Sci. Technol. Adv. Mater. 20 (2019) 710–724. doi: 10.1080/14686996.2019.1627174
Figure 1
Preparation of DHA-TPP NPs and proposed anti-tumor mechanism. The fabricated NPs can be accumulated in the tumor site through the EPR effect and internalized by tumor cells. Subsequently, DHA-TPP NPs are targeting mitochondria, thereby inducing mitochondrial dysfunction and activating the apoptosis pathway.
Figure 2
Characterization of DHA-TPP NPs. (A, B) Representative TEM images of DHA-TPP NPs. Scale bars: 500 nm (A) and 100 nm (B). (C) Particle size of DHA-TPP NPs. (D) Zeta-potential of DHA-TPP NPs. Data are expressed as mean ± standard deviation (SD) (n = 3).
Figure 3
Cellular uptake analysis. (A) Fluorescent images after incubating HepG2 cells with Cy5.5, DHA-TPP@Cy5.5 for 0, 1, 3, 6 h. (B, C) Flow cytometry analysis of cellular uptake capability. Data are expressed as mean ± SD (n = 3), ***P < 0.001.
Figure 4
Anti-tumor activity against HepG2 cells. (A) HepG2 viability after incubation with various concentrations of DHA and DHA-TPP NPs for 48 h. (B) Calcein-AM/PI staining of HepG2 cells after treatment with DHA and DHA-TPP NPs for 24 h, respectively. Data are expressed as mean ± SD (n = 3).
Figure 5
Anti-tumor mechanism analysis. (A) Determination of intracellular ROS contents in HepG2 cells with different treatments at 12 h (Scale bar: 100 µm). (B) Flow cytometric analysis of ROS generation in HepG2 cells with different treatments at 12 h. (C) Annexin V-FITC apoptosis detection in different samples. (D) Western blot analysis of Bax, cytochrome C, caspase-9 and Bcl-2 in HepG2 cells after treatment with 20 µmol/L DHA and DHA-TPP NPs for 24 h. (E) semi-quantitative analysis of Bax, cytochrome C, caspase-9 and Bcl-2 expression levels. Data are expressed as mean ± SD (n = 3), ***P < 0.001. GAPDH, recombinant glyceraldehyde-3-phosphate dehydrogenase.
Figure 6
Biodistribution of DHA-TPP NPs. (A) Representative in vivo fluorescence images of tumor-bearing mice at 2, 6, 12 and 24 h using ICG, ICG-labelled DHA-TPP NPs. (B) Semi-quantitative analysis of the fluorescence intensity in a tumor at designated time points after indicated treatments. (C) Ex vivo fluorescence images and (D) semi-quantitative of major organs from H22 tumor-bearing mice at 24 h. Data are expressed as mean ± SD (n = 3). **P < 0.01, ***P < 0.001.
Figure 7
Anti-tumor analysis in vivo. (A) Schematic illustration of in vivo pharmacodynamic study. (B) Tumor volume growth curves for different groups of mice after various treatments. (C) Photographs of excised tumors. (D) Average ex vivo tumor weights. (E) Body weight changes in tumor-bearing mice. (F) H&E, Ki67 and terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL) staining of tumor tissues after various treatments. Data are expressed as mean ± SD (n = 5), ***P < 0.001.

 DownLoad:
DownLoad:

 下载:
下载:








 下载:
下载:
