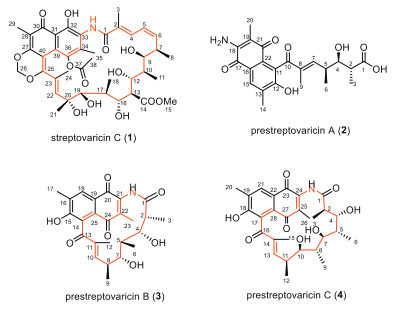
Figure 1.
Chemical structures of streptovaricin C (1) and prestreptovaricins A–C (2–4).
Premature termination in streptovaricin biosynthesis reveals unnatural short-chain analogues
Zi-Ru Wang , Pei-Yi Liu , Chao Zhang , Zhang-Yuan Yan , Hui-Ming Ge , Ren-Xiang Tan , Zi-Fei Xu

Ansamycins are a major family of macrolactam antibiotics characterized by an aromatic core, typically a benzene or naphthalene ring, bridged by a long aliphatic ansa chain [1,2]. These macrolactams are constructed by modular type Ⅰ polyketide synthases (PKSs) using 3-amino-5-hydroxybenzoic acid (AHBA) as the starter unit, followed by various post-PKS modifications [3]. Ansamycins include several pharmaceutically important agents, such as the clinically used anti-tuberculosis drug rifamycin SV [4], the antibody-drug conjugate kadcyla maytansinoid [5], and the Hsp90 inhibitor geldanamycin [6,7]. Among them, streptovaricins represent a unique subclass of naphthalenic ansamycins with potent activity against Mycobacterium tuberculosis and other Gram-positive pathogens, including methicillin-resistant Staphylococcus aureus (MRSA) [8-12].
The structural diversity of streptovaricins arises mainly from post-PKS tailoring modifications on the naphthalene ring and ansa bridge. Reported variations include hydroxylations at C-20 and C-28, dehydrogenation at C-21, and oxidation at Me-24 [12]. More unusual analogues comprise ansa ring-cleaved derivatives such as streptovaricin U and ansavaricins A, B, F-I, as well as ring-fused congeners such as ansavaricins C, E, and J [13-16]. In addition, damavaricins C and D and protostreptovaricins I-V have been isolated as plausible biosynthetic intermediates [17,18]. Despite this structural diversity, all known streptovaricins possess a C23-membered ansa chain, consistent with the long-standing view that type Ⅰ PKSs enforce strict chain-length fidelity [12].
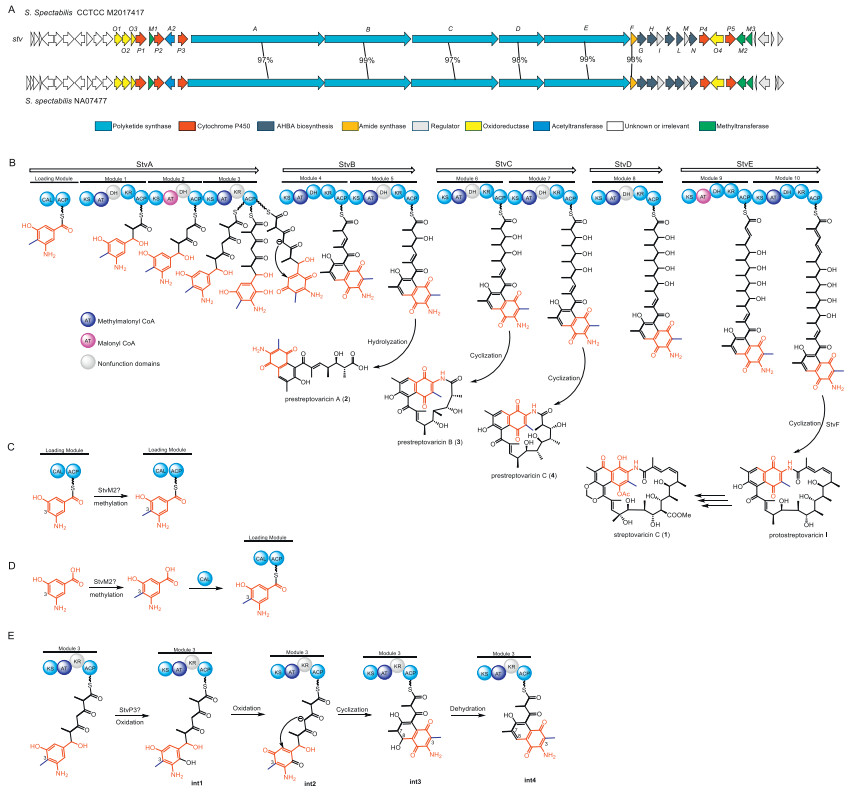
Recent biosynthetic studies have provided a detailed picture of streptovaricin assembly. The stv gene cluster from Streptomyces spectabilis CCTCC M2017417 encodes a modular PKS that incorporates the AHBA starter unit and extends it through ten cycles before macrolactamization to yield the C23 scaffold [12]. Notably, the macrolactamization step is distinctive among ansamycins, relying on a discrete arylamine N-acetyltransferase (NAT) family amide synthase rather than a canonical thioesterase (TE) domain for chain termination [19-21]. Cytochrome P450 monooxygenases stvP1, stvP4, and stvP5 catalyze hydroxylations at C-20, Me-24, and C-28, respectively, while stvP2 constructs the characteristic methylenedioxy bridge (MDB) [10,17]. Formation of the MDB is preceded by C6-O-methylation and C4-O-acetylation, catalyzed by stvM1 and stvA2, respectively [22]. Together, this enzymatic framework has reinforced the paradigm that streptovaricin biosynthesis is constrained to the C23 chain length.
In our ongoing efforts to discover novel secondary metabolites from actinomycetes, we identified Streptomyces spectabilis NA07477, which produces the canonical C23-membered streptovaricin C (1) along with three previously unreported metabolites: the C13 linear polyketide prestreptovaricin A (2) and the C15- and C17-membered macrolactams prestreptovaricins B (3) and C (4) (Fig. 1). To our knowledge, these represent the first naturally occurring streptovaricin congeners deviating from the strict C23 framework, thereby revealing unexpected plasticity in PKS chain-length control. Herein, we report the isolation, structural elucidation, and biosynthetic characterization of these unprecedented short-chain congeners, which provide new insights into the flexibility of ansamycin biosynthesis.
The molecular formula of the major metabolite, compound 1, was determined as C40H51NO14 by electrospray ionization high resolution mass spectrometry (ESI-HRMS) (Table S4 in Supporting information), identical to that of streptovaricin C. Its structure was fully established as streptovaricin C based on comprehensive nuclear magnetic resonance (NMR) analysis (including 1H, 13C NMR) and mass spectrometry data (Fig. 1 and Figs. S4–S6 in Supporting information) [15,23]. High performance liquid chromatography-mass spectrometry (HPLC-MS) analysis of the fermentation broth revealed the presence of compounds 2–4, which showed UV profiles similar to 1. Notably, their molecular weights were substantially lower than those of known streptovaricins, indicating that they are not simple analogues. This prompted us to isolate and purify 2–4 for further structural elucidation.
The molecular formula of prestreptovaricin A (2) was assigned as C22H25NO7 by ESI-HRMS. 1H and 13C NMR spectra displayed key signals including an aromatic singlet (δH 7.74), two exchangeable protons (δH 6.66, 9.75), an olefinic proton (δH 6.01), an aromatic methyl (δH 2.28), three singlet methyls (δH 1.75, 1.83, 2.28), two doublet methyls (δH 0.80, 0.90), and two methines (Table S5 in Supporting information). 1H–1H correlation spectroscopy (COSY) correlations defined a spin system between H-2 and H-7, including, including two branched methyls and a hydroxyl (4-OH). Heteronuclear multiple bond correlation (HMBC) data confirmed a polyketide-derived chain. Key correlations from H-9 to C-7, C-8, and C-10 established the C1-C10 branch. Additional HMBC correlations (H3–14 to C-12/C-13/C-15; H3–20 to C-18/C-19/C-21; H-15 to C-13/C-17), along with chemical shifts of C-11-C-22, revealed a naphthoquinone moiety (Fig. 2). The NH2 group was linked to C-18 via HMBC, and connectivity between C-10 and C-11 was deduced from molecular weight, completing the planar structure. The observed signal splitting is attributed to quinone-phenol tautomerism of the naphthoquinone core in solution, which is common for such structures. For clarity in reporting, the NMR data correspond to the dominant tautomeric form (Table S5 in Supporting information).
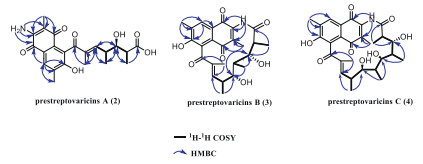
Prestreptovaricin B (3) had the molecular formula C25H29NO7 by ESI-HRMS. NMR signals included an aromatic singlet (δH 7.73), two exchangeable protons (δH 5.12, 6.63), an olefinic proton (δH 5.98), six methyls (δH 2.26, 1.89, 1.75, 1.16, 0.76, 0.68), and five methines (δH 4.09, 3.59, 2.76, 2.20, 2.02) (Table S6 in Supporting information). 1H–1H COSY revealed two spin systems: between C2 and C6, and between C7 and C10, connected via C-5 and C-7 based on HMBC (H3–6 to C-4/C-7; H-4 to C-7; H-7 to C-4). HMBC from H3–12 to C-10/C-11/C-13 indicated methylation at C-11. Two oxygenated methines (δC 68–84) were observed. Additional HMBC correlations (H3–17 to C-15/C-16/C-18; H3–23 to C-21/C-22/C-24; H-18 to C-15/C-19) and chemical shifts supported a naphthoquinone unit (Fig. 2). The amide NH showed HMBC to C-20, C-21, and C-22, confirming the attachment to the polyketide chain via an amide bond, thereby establishing the planar structure (Fig. 1).
Prestreptovaricin C (4) was assigned the molecular formula C28H35NO8 by ESI-HRMS. NMR features included an aromatic singlet (δH 7.73), two exchangeable protons (δH 6.63, 4.35), an olefinic proton (δH 6.10), an aromatic methyl (δH 2.27), a singlet methyl (δH 1.73), four doublet methyls (δH 1.88, 0.85, 0.73, 0.73), a triplet methyl (δH 1.01), and seven methines (Table S7 in Supporting information). 1H–1H COSY defined an extended spin system between H-2 and H-13, including four branched methyls and a hydroxyl (10-OH). HMBC data supported a polyketide chain (Fig. 2). Comparison with 3 indicated a similar naphthoquinone core. Key HMBC from NH to C-23 and C-25 confirmed the amide linkage between the chain and the naphthalene unit, completing the planar structure (Fig. 1).
Biosynthetic analysis indicated that analogues 2–4 originate from the same PKS gene cluster as the co-produced streptovaricin C (1). As shown in Fig. 3, in modular PKS pathways (Fig. 3B), the stereochemical outcome is strictly dictated by the high fidelity of tailoring domains, particularly the ketoreductase (KR) domains that set the configuration of each hydroxyl–bearing stereocenter [24]. Since 2–4 are congeners assembled by the same upstream modules as 1, they are expected to share identical configurations at their conserved stereocenters. This approach, assigning stereochemistry based on biosynthetic relatedness, represents a well-established practice for structure elucidation of novel natural product analogues (Fig. 1) [25-27]. Prestreptovaricin B (2) possesses a C15-ansa chain; whereas the only previously reported naphthalenic ansamycins with this feature are rubradirin and protorubradirin [17]. Notably, prestreptovaricin C (3) contains an unprecedented C17 ansa chain, a feature not previously observed in any ansamycin. Most significantly, these metabolites represent the first deviation from the prototypical C23 framework in the streptovaricin family. The newly isolated 2–4 were evaluated for antibacterial and cytotoxic activities. None exhibited significant activity (minimum inhibitory concentration (MIC) > 128 µg/mL) against Gram-positive (Staphylococcus aureus ATCC 6538 and Bacillus subtilis ATCC 9372) or Gram-negative (Pseudomonas aeruginosa ATCC 27853 and Escherichia coli ATCC 25922) strains. Likewise, no cytotoxicity was detected against the human cancer cell lines HepG2 and SW1990 at the tested concentrations (half maximal inhibitory concentration (IC50) > 50 µmol/L). The lack of activity is likely attributable to the absence of post-PKS modifications, consistent with previous structure-activity relationship studies of streptovaricin analogues [12].
Although the lack of post-PKS modifications is a plausible explanation for the loss of bioactivity in 2–4, the shortened ansa chain length itself may also contribute to their inactivity. Previous structure-activity relationship (SAR) studies have indicated that the C23 ansa chain is critical for optimal binding to biological targets, such as RNA polymerase in the case of rifamycins, or Hsp90 in geldanamycin analogues. The reduced ring size in 3 (15-membered) and 4 (17-membered) likely alters the spatial conformation and flexibility of the molecule, thereby impairing its ability to interact effectively with cellular targets. Thus, both the absence of essential tailoring modifications and the non-canonical chain length may synergistically account for the observed lack of antibacterial and cytotoxic activities.
In polyketide (PKS) and nonribosomal peptide (NRPS) biosynthetic pathways, synthesis often deviates from strict linear modularity [24]. Intermediates can prematurely dissociate from the assembly line, yielding truncated products, or bypass specific catalytic modules via skipping mechanisms [28,29]. Such premature release or module skipping increases structural diversity in secondary metabolites and highlights the flexibility of these pathways [30-32].
To rationalize the formation of macrolactams 3 and 4, we first performed a retrobiosynthetic analysis to challenge the module-skipping hypothesis. The canonical C23 streptovaricin assembly line comprises ten PKS modules, culminating in macrolactamization of the full-length chain by the dedicated amide synthase, stvF. However, the structures of the truncated analogues are inconsistent with this pathway. Specifically, the C15 backbone of 3 is devoid of all structural features installed by modules 8, 9, and 10, including the characteristic C-20 hydroxyl and C-24 methyl groups. Likewise, the C17 chain of 4 lacks the modifications introduced by modules 9 and 10. A module-skipping scenario would require the terminal enzymatic machinery, namely, the ACP domain of module 10 (ACP10) and the amide synthase stvF, to recognize and process a non-native, truncated substrate from an upstream module (e.g., module 6 or 7). Such an event is mechanistically improbable due to the stringent substrate specificity these domains exhibit for their cognate, full-length polyketide precursors. Therefore, while module skipping can occur in some PKS systems, the structural evidence here argues strongly against it, favoring premature chain release as the operative mechanism.
We hypothesize that 2–4 arise from such non-canonical events. Specifically, C13 linear polyketide 2 likely results from hydrolysis after elongation to module 5, whereas C15 and C17 macrolactams 3 and 4 could be generated either through amide synthase-mediated release after elongation to modules 6 and 7, respectively (Fig. 3B). To test these hypotheses, we first sought a genetically tractable surrogate strain, since conjugation-based gene disruption was intractable in NA07477. Phylogenetic analysis of the core PKS and amide synthase genes revealed > 90% sequence identity between the stv clusters of NA07477 and CCTCC M2017417 (Fig. 3A). Consistently, CCTCC M2017417 also produced 2–4 (Fig. S3 in Supporting information), validating it as a suitable host for subsequent functional studies. The 16S rRNA gene sequence of Streptomyces spectabilis NA07477 has been deposited in GenBank under accession number PPX463749. The genome sequence of the surrogate strain Streptomyces spectabilis CCTCC M2017417 is publicly available under Assembly accessionnumber GCA_000785105.
To elucidate the mechanism of chain termination, we conducted targeted site-directed mutagenesis and gene inactivation experiments (Figs. S1 and S2 in Supporting information). First, to test the module skipping hypothesis, the ACP domains of the terminal PKS modules 9 (stvE-ACP1) and 10 (stvE-ACP2), as well as the entire module 8 (stvD), were inactivated. None of these mutations abolished production of compounds 2–4 (Fig. 4), effectively ruling out a late-stage module skipping mechanism. In contrast, mutations in earlier modules directly affected the production of the shortened congeners. Inactivation of the module 7 ACP (stvC-ACP2) completely abrogated the production of the C17 macrolactam 4 (Fig. 4, trace iii). More definitively, inactivation of the module 6 ACP (stvC-ACP1) abolished both 3 and 4, leaving only the C13 linear product 2 (Fig. 4, trace ii). These results provide strong evidence that 3 and 4 arise from premature chain release and cyclization at modules 6 and 7, respectively, while 2 represents the hydrolysis product of the module 5 intermediate.
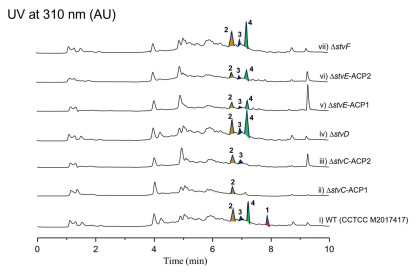
Finally, we investigated whether the dedicated amide synthase stvF was responsible for premature cyclization. Unexpectedly, knockout of stvF gene did not abolish the production of the cyclized 3 and 4 (Fig. 4, trace vii), suggesting that another promiscuous cyclase encoded elsewhere in the genome may catalyze this premature macrolactamization. This prompts us to consider non-canonical mechanisms for macrolactamization, potentially involving a Type Ⅱ thioesterase (TEII) or even a condensation (C) domain from a separate NRPS system, known for their ability to process aberrant intermediates [33-35]. The potential involvement of such a distally encoded, non-homologous enzyme presents a significant challenge to its identification via conventional genome mining.
While premature chain termination is a recognized source of diversity in modular PKSs, leading to truncated analogues in pathways like those of rifamycin [21], erythromycin [36], and tetrafibricin [37], our findings reveal a crucial distinction. In many previously reported cases, prematurely released chains are simply hydrolyzed to their linear forms or their cyclization still relies on the canonical terminal domain of the PKS [34,38]. In sharp contrast, the intermediates leading to 3 and 4 are efficiently intercepted and cyclized by an enzymatic activity completely independent of the primary assembly line. Therefore, this stvF-independent macrolactamization uncovers a previously unrecognized mechanism for chain release and diversification in ansamycin biosynthesis, highlighting a new mode of metabolic crosstalk. The elucidation of this elusive catalytic activity is a primary objective of our ongoing research.
While significant progress has been made in understanding streptovaricin biosynthesis, several key steps remain unclear, including the C-3 methylation of AHBA and the formation of the naphthalene ring. The observation that the C-3 methyl group is already present in the prematurely released congeners 2–4 strongly suggests that methylation occurs early in the pathway. The C-3 methylation of AHBA is likely to occur either during the early stages of PKS assembly (Fig. 3C) or prior to incorporation by the loading module (Fig. 3D). This is analogous to the biosynthesis of chaxamycins, where the methyltransferase Cxm24 is proposed to catalyze the C-3 methylation of AHBA [39]. Bioinformatics analysis of the stv BGC revealed StvM2 as a close homolog of Cxm24 (78% identity), implicating it as the most likely candidate for this reaction.
The structures of intermediates 2–4 provide compelling evidence that the naphthalene core is established and aromatized at an earlier PKS stage than previously assumed. Based on this, we propose an updated pathway initiated by the P450 stvP3 (Fig. 3E), a conserved and essential enzyme in naphthalenic ansamycin biosynthetic pathways [21,40]. This hypothesis is strongly supported by previous genetic studies, which demonstrated that the knockout of stvP3 abolished product formation, a phenotype that was fully restored upon gene complementation [12]. These findings suggest stvP3 acts at an early stage, likely catalyzing a key hydroxylation on a PKS-bound intermediate that triggers the subsequent cyclization and aromatization cascade (Fig. 3E).
However, the precise mechanism, sequence of enzymatic events, and all proposed intermediates in this pathway (Fig. 3E) remain hypothetical and await experimental verification. Definitive validation is exceptionally challenging, as it requires the in vitro reconstitution of this on-line PKS processing step. Our future work will therefore focus on this goal by expressing and purifying stvP3. We then plan to test its activity against a synthetic polyketide intermediate, attached to either the module 3 ACP or a carrier protein mimic (e.g., N-acetylcysteamine, SNAC), and monitor for the formation of the key reaction intermediates.
In summary, we have identified and characterized three unprecedented shortened streptovaricin congeners, prestreptovaricins A–C, representing the first naturally occurring deviations from the canonical C23 ansa framework. Genetic and mutagenesis studies demonstrated that these metabolites arise from premature chain termination at modules 5–7, followed by macrolactamization, rather than from module skipping. Moreover, gene knockout experiments suggest that alternative cyclases outside the stv gene cluster can mediate premature ring closure, highlighting unexpected flexibility in the chain release step of ansamycin biosynthesis. Together, these findings expand current understanding of PKS chain-length control and provide new insights into the enzymatic logic underlying structural diversification in ansamycins.
The authors declare that they have no known competing finan cial interests or personal relationships that could have appeared to influence the work reported in this paper.
Zi-Ru Wang: Writing – original draft, Resources, Investigation, Funding acquisition, Formal analysis. Pei-Yi Liu: Visualization. Chao Zhang: Software. Zhang-Yuan Yan: Resources. Hui-Ming Ge: Funding acquisition, Formal analysis, Data curation, Conceptualization. Ren-Xiang Tan: Resources, Funding acquisition, Conceptualization. Zi-Fei Xu: Writing – review & editing, Writing – original draft, Investigation.
We are indebted to Prof. Yuhui Sun (Huazhong University of Science and Technology) for providing S. spectabilis CCTCC M2017417. This work was financially supported by Ministry of Science and Technology (No. 2022ZD0211800), National Natural Science Foundation of China (Nos. 82525108, 22193071, 22437003, W2412037, 22377052 and 22507128) and the Natural Science Foundation of Yunnan (No. 202501CF070088).
Supplementary material associated with this article can be found, in the online version, at doi:
V. Prelog, W. Oppolzer, Helv. Chim. Acta 25 (1973) 1179–1187.
W. Wehrli, Top. Curr. Chem. 72 (1977) 21–49.
Q. Kang, Y. Shen, L. Bai, Nat. Prod. Rep. 29 (2012) 243–263. doi: 10.1039/C2NP00019A
N. Maggi, C.R. Pasqualucci, R. Ballotta, et al., Chemotherapy 11 (1966) 285–292. doi: 10.1159/000220462
J.M. Cassady, K.K. Chan, H.G. Floss, et al., Chem. Pharm. Bull. 52 (2004) 1–26.
L. Whitesell, E.G. Mimnaugh, B. DeCosta, et al., Proc. Natl. Acad. Sci. U. S. A. 91 (1994) 8324–8328. doi: 10.1073/pnas.91.18.8324
R.X. Wang, R.T. Zhang, H.R. Yang, et al., Chin. Chem. Lett. 34 (2023) 107529. doi: 10.1016/j.cclet.2022.05.043
P. Siminoff, R.M. Smith, W.T. Sokolski, et al., Am. Rev. Tuberc. 75 (1957) 576–583.
S. Mizuno, H. Yamazaki, K. Nitta, et al., Biophys. Acta Nucleic Acids Protein Synth. 157 (1968) 322–332.
A. Ravina, Presse. Med. 65 (1957) 834.
J.S. Horoszewicz, K.L. Rinehart Jr., S.S. Leong, et al., Antimicrob. Agents Chemother. 7 (1975) 281–284. doi: 10.1128/AAC.7.3.281
Y. Liu, X. Chen, Z. Li, et al., ACS Chem. Biol. 12 (2017) 2589–2597. doi: 10.1021/acschembio.7b00467
W.M. Knoll, K.L. Rinehart, Jr., P.F. Wiley, et al., J. Antibiot. 33 (1980) 249–251. doi: 10.7164/antibiotics.33.249
Z.Q. Zhang, X.K. Wu, R.T. Song, et al., RSC Adv. 7 (2017) 14857–14867. doi: 10.1039/C7RA00961E
Z.Q. Zhang, J.L. Zhang, R.T. Song, et al., RSC Adv. 7 (2017) 5684–5693. doi: 10.1039/C6RA27405F
Y.Z. Liu, X. Chen, Z.Y. Li, et al., Chem. Biodivers. 17 (2020) e1900713. doi: 10.1002/cbdv.201900713
K.L. Rinehart Jr., F.J. Antosz, P.V. Deshmukh, et al., J. Antibiot. 29 (1976) 201–203. doi: 10.7164/antibiotics.29.201
P.V. Deshmukh, K. Kakinuma, J.J. Ameel, et al., J. Am. Chem. Soc. 98 (1976) 870–872. doi: 10.1021/ja00419a056
W. Ewert, C. Bartens, J. Ongouta, M. Holmes, et al., Nat. Commun. 16 (2025) 2464.
F. Pompeo, A. Mushtaq, E. Sim, Protein Expr. Purif. 24 (2002) 138–151. doi: 10.1006/prep.2001.1550
T.W. Yu, Y. Shen, Y. Doi-Katayama, et al., Floss. Proc. Natl. Acad. Sci. U. S. A. 96 (1999) 9051–9056. doi: 10.1073/pnas.96.16.9051
G. Sun, C. Hu, Q. Mei, et al., Nat. Commun. 11 (2020) 4501. doi: 10.1038/s41467-020-18336-5
K.L. Rinehart Jr., H.H. Mathur, K. Sasaki, et al., J. Am. Chem. Soc. 90 (1968) 6241–6243. doi: 10.1021/ja01024a067
C. Hertweck, Angew. Chem. Int. Ed. 48 (2009) 4688–4716. doi: 10.1002/anie.200806121
J. Cui, P.F. Hillman, G.J. Kim, et al., Nat. Prod. Rep. 42 (2025) 1136–1174. doi: 10.1039/d4np00061g
J. Feng, J. Liu, T. Ye, Org. Lett. 27 (2025) 9299–9303. doi: 10.1021/acs.orglett.5c02933
I.R.G. Thistlethwaite, F.M. Bull, C. Cui, et al., Chem. Sci. 8 (2017) 6196–6201. doi: 10.1039/C7SC01670K
D.C. Wang, H.J. Mao, Z.L. Zhao, et al., Adv. Sci. 11 (2024) 2401708. doi: 10.1002/advs.202401708
T. Awakawa, M. Crüsemann, J. Munguia, et al., ChemBioChem. 16 (2015) 1443–1447. doi: 10.1002/cbic.201500177
G. Zhai, W. Wang, W. Xu, G. Sun, et al. Angew. Chem. Int. Ed. 59 (2020) 22738–22742. doi: 10.1002/anie.202011357
M. Herisse, K. Ishida, J. Staiger-Creed, et al. ACS Chem. Biol. 18 (2023) 1872–1879. doi: 10.1021/acschembio.3c00311
S.C. Wenzel, B. Kunze, G. Höfle, et al., ChemBioChem 6 (2005) 375–385. doi: 10.1002/cbic.200400282
M. Kotowska, K. Pawlik, Appl. Microbiol. Biotechnol. 98 (2014) 7735–7746. doi: 10.1007/s00253-014-5952-8
F. Kopp, M.A. Marahiel, Nat. Prod. Rep. 24 (2007) 735–749. doi: 10.1039/b613652b
F.L. Jiang, A. Liu, Q. Wei, et al., Chin. Chem. Lett. 35 (2024) 109504. doi: 10.1016/j.cclet.2024.109504
A.F.A. Marsden, P. Caffrey, J.F. Aparicio, et al., Science 263 (1994) 378–379. doi: 10.1126/science.8278811
L.H. Zhang, T. Hashimoto, B. Qin, et al., Angew. Chem. Int. Ed. 56 (2017) 1740–1745. doi: 10.1002/anie.201611371
D.L. Akey, J.D. Kittendorf, J.W. Giraldes, et al., Nat. Chem. Biol. 2 (2006) 537–542. doi: 10.1038/nchembio824
J.F. Castro, V. Razmilic, J.P. Gomez-Escribano, et al., Appl. Environ. Microbiol. 81 (2015) 5820–5831.
H.G. Floss, T. W, Yu, Chem. Rev. 105 (2005) 621–632. doi: 10.1021/cr030112j

Figure 1 Chemical structures of streptovaricin C (1) and prestreptovaricins A–C (2–4).

Figure 3 (A) Genetic organization of the stv cluster from S. spectabilis CCTCC M2017417 or S. spectabilis NA07477. (B) Proposed biosynthetic pathway leading to 1–4. Two possible routes for C-3 methylation of the AHBA: (C) on-line methylation at the ACP-tethered AHBA, (D) pre-loading methylation of free AHBA. (E) Proposed route for the formation of the naphthalene ring. The abbreviations of each domain designations as CAL, carboxylic acid: ACP ligase; KS, β-ketoacyl-ACP synthase; AT, acyltransferase for loading a malonyl-CoA or a methylmalonyl-CoA; DH, β-hydroxyacyl-thioester dehydratase; KR, β-ketoacyl-ACP reductase; ACP, acyl carrier protein.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:


 下载:
下载: