Scheme 1.
Three-step synthesis of pillarrhombimine PIm, proton allocation based on 1H and NOESY NMR, and structures of nitrile guests G1-G5.
Imination enables efficient formation of a pillararene-inspired host with endo-cavity hydrogen-bonding capability
Sem Bleus , Sergio Ribone , Renny Maria Losus , Liliana Dobrzańska , Luc Van Meervelt , Wim Dehaen

Since Pedersen's discovery of crown ethers and their metal ion binding [1], an expanding number of macrocycles with preorganized cavities and suitable host-guest properties has been developed [2-7]. Among these macrocycles, methylene-linked [1n] cyclophanes, such as cyclotriveratrylenes [8], calix[n]arenes [9] and resorcin[n]arenes [10], have garnered significant attention due to their deep π-cavities, which allow for enveloping guests. Pillar[n]arenes (Pns), composed of cyclic para methylene-bridged hydroquinone arrays, represent a relatively recent addition to this receptor class [11]. Their synthetic accessibility, ease of functionalization and rigid, highly symmetrical, electron-rich cavities render them able to act as hosts to neutral, electron-deficient guests, which has seeded potential applications ranging from biology to materials science [12-16]. These benefits have enticed considerable research into related symmetry-reduced and/or bridge-functionalized pillar[n]arene-inspired arenes (PIAs) of diverse size, geometry and guest affinity [17,18]. Incorporating these conformational and supramolecular traits typically results in low-yielding cyclizations and tedious product separation, sharply contrasting the straightforward multigram synthesis of the parent host.
A well-documented modification pathway for Pns recently gaining attention is the (partial) introduction of heteroatoms rather than methylenes as bridging units, leading to properties not readily achieved in Pns. In 2024, Ogoshi and coworkers reported silapillar[n]arenes [19], showing efficient σ*−π* conjugation, while the Ogoshi and Sue groups independently developed methods toward azapillar[6]arenes, which enabled selective belt functionalization [7,20]. Furthermore, structures incorporating oxygen [21], sulfur [22,23], disulfide [24] and sulfur-containing polyatomic bridges [25] have been prepared, portraying the generality of the method to create chemical space in Pn hosts. Nonetheless, apart from the solid-state adsorption of iodine [7], these structures have not been reported to afford guest binding. Introducing similar bridges in calix[n]arenes [26] and dialkoxynaphthalene macrocycles [27] has contrastingly evoked the binding of guests distinct from those typically displaying affinity to their all-carbon analogs. In this regard, heterapillar[n]arene-type macrocycles enhancing the chemical space while enabling guest recognition complementary to the parent host are desired.
Based on this actuality within the research field and interest in homoheteracyclophanes [24,28], we aimed to develop an efficient, scalable, chromatography-free route to a new PIA possessing a suitable cavity for accommodating guests. As cyclization strategy, dynamic covalent chemistry, specifically imination, was chosen based on several factors. Firstly, its reversibility can produce covalent self-assemblies in excellent yield without requiring byproduct removal [29-32] by eliminating kinetic side-products [33]. Secondly, imination has not been widely investigated to provide PIAs [34], despite extensive literature on the successful construction of imine-containing hosts [35-38]. Thirdly, endo-cavity hydrogen bonding is considered challenging to achieve in macrocyclic arenes [39] but could be facilitated by the introduced nitrogen atoms. Therefore, we report the synthesis, structure and host-guest behavior of pillarrhombimine, a tetraimine PIA, and its modification to its amine analog. Combining imines with 1,4-dialkoxybenzenes is envisioned to promote neutral molecule recognition over related imines displaying limited guest binding [40] or only recognizing metal ions [41]. Additionally, a larger cavity than for pillar[3]trianglimine [34] should facilitate binding guests beyond linear alkanes.
A fragment coupling approach was adopted to synthesize a suitable imine macrocycle (Scheme 1). The key dimer 4 was prepared in 71% overall yield by the Friedel Crafts alkylation of benzyl alcohol 1 with an excess 1,4-dimethoxybenzene 2, followed by Rieche formylation. When reacting equimolar amounts of the resulting dialdehyde 4 and 1,2-diaminoethane (EDA) in DCM at room temperature (Scheme 1), mass spectrometry revealed near-complete conversion to the [2 + 2] pillarrhombimine PIm (Fig. S7 in Supporting information). Solvent evaporation quantitatively afforded the pure macrocycle, sharply contrasting the tedious separation of undesired oligomers for other Pn-inspired hosts. The synthesis could be scaled up to 3 mmol without performance loss, rendering the reaction scale and overall yield among the highest within PIAs. The macrocyclization yield is significantly improved over similarly-sized PIAs prepared by Friedel-Crafts alkylation (i.e., helic[1]triptycene[3]arene (2.5-fold), leggero pillar[5]arene (3-fold), triptycene-embedded pillararene (16-fold)) or nucleophilic substitution strategies (ProBox (5-fold), CH2S-pillar[4]arene (8-fold)) [15]. The three-step process is comparable in yield to the optimized pillar[5]arene synthesis by Ogoshi et al. [42] and the macrocyclization is slightly more efficient than the highest reported yields for pillar[5]arene [43]. The reaction was driven to completion in dry or untreated solvent, indicating a high thermodynamic driving force for cyclization. As performing the reaction in chloroform did not alter the reaction efficiency, the covalent self-assembly seems to stem from self-templation [44].
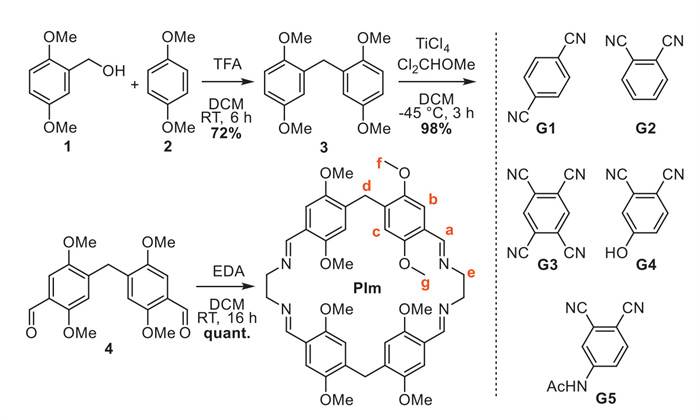
X-ray suitable single crystals of PIm (CCDC: 2381796) in the monoclinic space group P21/c were obtained by slow room temperature evaporation from a DCM/heptane mixture, unambiguously confirming [2 + 2] adduct formation (Fig. 1). The macrocycle adopts a rhombic geometry with three aryl units perpendicular to the cavity plane and a single inwardly tilted one stabilized through C—H⋯O short contacts with adjacent units (Fig. 1b, Fig. S14 and Table S3 in Supporting information). PIm possesses a racemic mixture of planar chiral enantiomers (pSSSS and pRRRR) resulting from the methoxy group orientation. These enantiomers combine into solid-state dimers by occupying the cavity of one another with a single methoxy group (Fig. 1b), which stack into infinite 1D polymeric motifs along the a-axis (Fig. 1c). A C—H⋯π, C—H⋯N and C—H⋯O hydrogen bonding network organizes these structures (Table S3). The van der Waals-corrected aryl-to-aryl distance is around 4.6 Å, highly similar to the 4.7 Å cavity size of permethoxypillar[5]arene (P5) [11]. The aryl-flanked methylene bridges are 6.1 Å apart and imine carbons of the same dimeric unit are distanced at 6.3 Å, slightly larger than P5 (5.7 Å) but significantly smaller than for pillar[6]arene (8.3 Å).
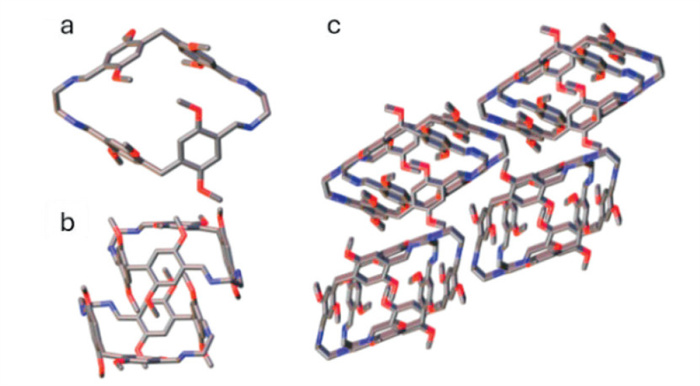
The solution-state conformations of PIm were assessed using NMR experiments and DFT calculations. The 1H NMR spectra of PIm only displayed singlet resonances for protons a-g (Scheme 1), implying a high symmetry and fast conformational interconversion at room temperature. Theoretical calculations confirmed the preferential adoption of a symmetrical, fully open conformation in solution, stabilized 14.5 kcal/mol more in chloroform than the solid-state conformation. In this state, the inner π-faces of the dimethoxybenzenes create a high electron density cavity, resembling P5 in electron richness (Figs. S31 and S32 in Supporting information). The nitrogen lone pairs produce additional electron-rich sites in the cavity vertices of interest toward hydrogen bonding. Variable temperature NMR indicated high flexibility as no signal splitting occurred for temperatures down to −89 ℃ (Fig. S13 in Supporting information). DFT calculations, in which the dihedral angle of a single 1,4-dimethoxybenzene was rotated in 5° increments relative to the cavity plane, corroborated the host flexibility (Fig. 2, Figs. S34, S38-S43 in Supporting information). A 6.8 kcal/mol rotation barrier (Ea) occurs when the methoxy group f occupies the cavity (−10°). Rotating the opposing group g inward induces a slightly lower barrier (6.6 kcal/mol, 185°) as the methoxy group experiences less steric hindrance upon passing the EDA unit. Equivalent calculations on P5 (Ea = 8.2 kcal/mol) revealed that PIm undergoes planar chiral inversion at a higher rate and represents a more flexible Pn analog (Fig. 2).
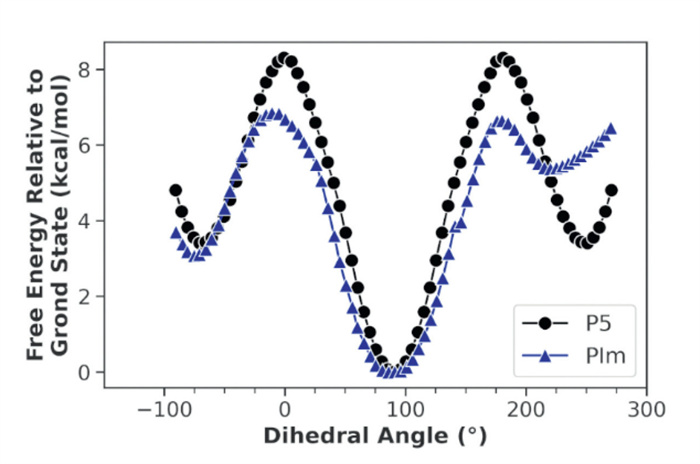
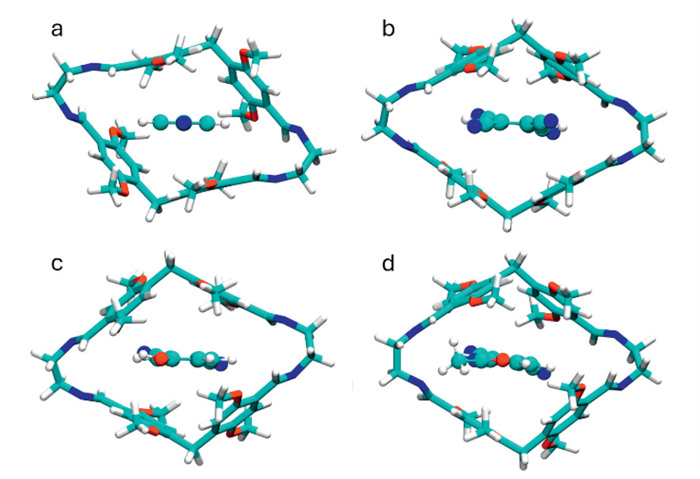
Due to its similarities to P5, PIm was evaluated as a host for neutral, electron-deficient species. Succinonitrile, adiponitrile and 1,3-dimethylimidazolium iodide, archetypical P5 guests [45,46], were probed but did not lead to observable interactions. The rhombic geometry of PIm led to affinity for terephthalonitrile G1, which is not recognized by Pns [47] and azapillar[6]arene [7], pointing at complementarity in guest recognition to both hosts. An evident upfield shift of methylene protons d under fast exchange relative to the NMR time scale upon guest addition implied guest inclusion. Complexation was confirmed by 2D NOESY NMR (Fig. S19 in Supporting information) and fitting of NMR titration data afforded a 1:1 binding constant (Ka) of 17 ± 3 L/mol. DFT calculations indicated that the host adopts a bowl-shaped geometry to partially embed G1 in a tilted conformation (Fig. 3a). Complex stabilization occurs through C—H⋯N interactions of a single nitrile with methoxy protons f and g, C—H⋯π and π⋯π interactions (Figs. S44 and S49 in Supporting information).
Based on the successful binding of G1, other cyanobenzenes G2-G5 were evaluated as guests. 1,2-Dicyanobenzene G2 showed no interaction, possibly due to the favored symmetrical guest positioning (Figs. 3b-d) increasing the distance to the host π-systems, diminishing C—H⋯π and π⋯π interactions compared to G1. Additionally, fitting the more sterically hindered aryl system in the cavity may disfavor guest binding.
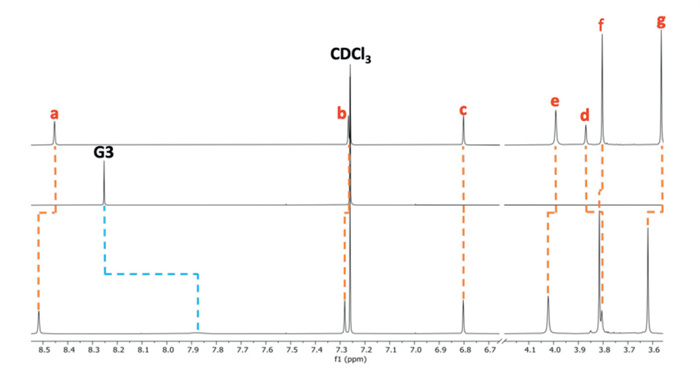
Conversely, 1H NMR suggested complexation with the more electron-deficient 1,2,4,5-tetracyanobenzene G3. Under fast exchange, considerable broadening and significant shielding from 8.25 ppm to 7.88 ppm of the G3 resonance was observed upon adding 1 equiv. of the host (Fig. 4). Guest encapsulation was supported by DOSY NMR, with the guest exhibiting a diffusion coefficient closely related to the free host (Fig. S20 in Supporting information) [48,49]. The binding strength was quantified by 1H NMR titration (Fig. S24 in Supporting information), affording a Ka of (5.0 ± 1.6) × 103 L/mol, a 180-fold improvement over pillar[6]arene [46]. Despite various attempts, no suitable crystals of the complex were obtained. However, DFT calculations demonstrated that the open host geometry is suitable for forming a threaded complex (Fig. 3b) stabilized by four C—H⋯N interactions involving the methoxy groups f (Fig. S45 in Supporting information) and C—H⋯π interactions (Fig. S50 in Supporting information).
Interestingly, 3,4-dicyanophenol G4 aryl resonances underwent noticeable 0.3–0.5 ppm upfield 1H NMR shifts upon adding equimolar PIm (Fig. S25 in Supporting information), despite being more electron-rich than the non-binding G2. DOSY NMR confirmed that guest complexation took place (Fig. S21 in Supporting information). Fitting of the binding isotherm led to a Ka of (1.3 ± 0.1) × 103 L/mol. The change in affinity compared to G2 suggested hydrogen bonding, which was corroborated by the appearance of a broadened phenolic signal absent in the free guest. DFT calculations confirmed the type of interaction as guest encapsulation is stabilized by O—H⋯O-Me and methoxy–to-nitrile C—H⋯N hydrogen bonding (Fig. S46 in Supporting information). To the best of our knowledge, guests hydrogen bonding to the alkoxy rims has not been reported in pillar[5]arene, highlighting the effect of the altered geometry on the interaction mode. Non-covalent interaction analysis (Fig. S51 in Supporting information) revealed that the electron-donating group reduces the strength of the dispersion interactions between the guest conjugated system and the host aryl groups compared to G3, explaining the reduced affinity.
To further probe the endo-cavity hydrogen capacities of PIm, acetamide analog G5 was evaluated as guest. A binding constant of (4.9 ± 1.2) × 102 L/mol was derived from the upfield shifting of the protons e adjacent to the imine and complexation was confirmed by DOSY NMR. Deshielding of the amide hydrogen by 0.48 ppm evidenced its involvement in hydrogen bonding. Based on DFT calculations, an N—H⋯N=C hydrogen bond, weaker than the hydrogen bond in G4@PIm (Fig. S52 in Supporting information), is preferred over N—H⋯O-Me hydrogen bonding to avoid steric clashing of the guest acetyl and host methoxy groups. Additional interactions, apart from C—H⋯π interactions, involve non-classic hydrogen bonds of the host methoxy and guest nitrile, carbonyl oxygen and acetyl CH3 groups (Fig. S47 in Supporting information).
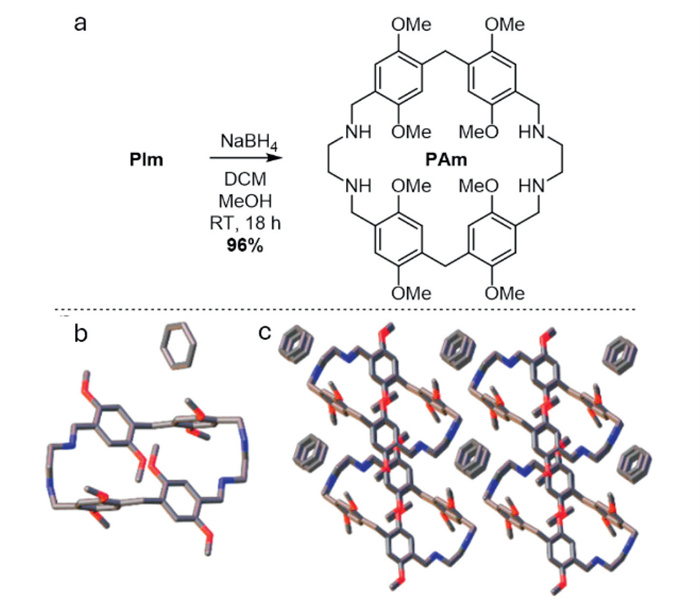
Finally, PIm was efficiently postfunctionalized by reduction to pillarrhombamine PAm (Scheme 2a). However, the guest binding capacity of the host seems markedly altered, as no change in 1H NMR shift occurred upon the addition of G1-G3, 2,6-dicyanonaphthalene or 1,3-dimethylimidazolium iodide. Single crystals of PAm (CCDC: 2382390) obtained by slow evaporation from benzene revealed a significant geometrical change. To form hydrogen bonds in the EDA units, two benzyl amines rotate out of the adjacent aryl plane. Resultantly, a rectangular molecular geometry without an intrinsic cavity is adopted (Scheme 2b). This rotation spaces two opposing aryl units and benzylamine methylenes belonging to the same dimeric unit at an increased 6.5 Å and 6.9 Å, respectively. The remaining aryl groups (4.4 Å) and the Ar-CH2-Ar bridges (4.7 Å) maintain a similar distancing as in PIm. The aryl groups possess a meso pRSRS planar chiral orientation and are tilted pairwise from the molecular plane. The arrangement of the macrocycles promotes forming a system of interconnected voids along the a-axis (Scheme 2c), which are occupied by solvent molecules. Each benzene is wedged between four macrocycles by C—H⋯π interactions (Table S3). The packing is stabilized through N—H⋯O and C—H⋯O short contacts. The intramolecular hydrogen bonds and the amine groups of PAm seem promising for binding hydrogen-bonding or salt-bridge forming guests [41]. These features may also facilitate stimuli-responsiveness toward solvents or pH, potentially leading to selective guest release and dynamic switching.
In conclusion, imination proves an excellent strategy to construct pillar[n]arene-like macrocycles with an altered geometry. Due to its reversible nature, the macrocycles can be synthesized straightforwardly without high purification requirements, contrasting various related macrocycles. The straightforward synthesis provides a clear benefit to applications, although the reversible nature might impact the host stability. The altered geometry affords a suitable functional group positioning to recognize di-, tri- and tetrasubstituted cyanobenzenes, with possible involvement of the host hydrogen bond acceptors. These guests are atypical for pillar[n]arenes, indicating complementarity of the novel receptor class. Reduction to the corresponding amine was conducted in high yield, leading to the introduction of hydrogen bond acceptors in the cavity plane. Future work will focus on expanding the modularity of the macrocyclic classes to overcome current limitations of Pns and PIAs.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Sem Bleus: Writing – review & editing, Writing – original draft, Methodology, Investigation, Formal analysis, Conceptualization. Sergio Ribone: Writing – review & editing, Software, Methodology, Investigation, Formal analysis. Renny Maria Losus: Writing – review & editing, Investigation, Formal analysis. Liliana Dobrzańska: Writing – review & editing, Validation, Supervision, Resources, Project administration, Funding acquisition, Formal analysis. Luc Van Meervelt: Writing – review & editing, Validation, Resources, Investigation, Funding acquisition, Formal analysis. Wim Dehaen: Writing – review & editing, Supervision, Resources, Project administration, Funding acquisition, Conceptualization.
This work was supported by the Research Foundation Flanders (FWO) [doctoral fellowship 11G8123, Weave G00124N, infrastructure grants I001920N & I002720N, and Scientific Research Community W000620N]; the KU Leuven [postdoctoral fellowship PDMT2/24/051]; the National Science Centre, Poland [OPUS call in the Weave program grant 2022/47/I/ST5/02127] and the Hercules Foundation of the Flemish Government [No. 20100225–7 and Project AKUL/09/0035]. The authors acknowledge Gert Steurs for assistance during NMR spectrometry, as well as Jef Rozenski and Lize Bynens for MS measurements. The authors thank the CCAD (
Supplementary material associated with this article can be found, in the online version, at doi:
C.J. Pedersen, J. Am. Chem. Soc. 89 (1967) 2495–2496. doi: 10.1021/ja00986a052
W. Zhang, W. Yang, J. Zhou, Molecules 28 (2023) 4422. doi: 10.3390/molecules28114422
S. Bai, L.W. Zhang, Z.H. Wie, et al., Nat. Commun. 15 (2024) 6616. doi: 10.1038/s41467-024-50980-z
F. Zeng, L.L. Tang, W.H. Bao, et al., Org. Chem. Front. 11 (2024) 3404–3408. doi: 10.1039/d4qo00407h
N. Liu, X.N. Han, H. Ma, et al., Org. Lett. 26 (2024) 7239–7243. doi: 10.1021/acs.orglett.4c02709
K. Zhang, X.Y. Lou, Y. Wang, et al., Chin. Chem. Lett. 36 (2025) 110464. doi: 10.1016/j.cclet.2024.110464
W. Fang, J. Zhang, M. Guo, et al., Angew. Chem. Int. Ed. 63 (2024) e202409120. doi: 10.1002/anie.202409120
M.J. Hardie, Chem. Soc. Rev. 39 (2010) 516–527. doi: 10.1039/B821019P
P. Neri, J.L. Sessler, M.X. Wang, Calixarenes and Beyond, Springer International Publishing, Cham, 2016.
D.J. Cram, J.M. Cram, Container Molecules and Their Guests, The Royal Society of Chemistry, Cambridge, 1994.
T. Ogoshi, S. Kanai, S. Fujinami, et al., J. Am. Chem. Soc. 130 (2008) 5022–5023. doi: 10.1021/ja711260m
T. Xiao, L. Zhou, L. Xu, et al., Chin. Chem. Lett. 30 (2019) 271–276. doi: 10.1016/j.cclet.2018.05.039
K. Kato, S. Fa, S. Ohtani, et al., Chem. Soc. Rev. 51 (2022) 3648–3687. doi: 10.1039/d2cs00169a
K. Wang, X. Tian, J.H. Jordan, et al., Chin. Chem. Lett. 33 (2022) 89–96.
T.H. Shi, S. Ohtani, K. Kato, et al., Trends. Chem. 5 (2023) 537–550. doi: 10.1016/j.trechm.2023.04.004
T. Ogoshi, X. Li, J. Yang, et al., Adv. Mater. 36 (2024) 2313317. doi: 10.1002/adma.202313317
S. Bleus, W. Dehaen, Coord. Chem. Rev. 509 (2024) 215762. doi: 10.1016/j.ccr.2024.215762
J. Jiao, J. Yu, X. Tian, et al., Chin. Chem. Lett. 36 (2025) 111026. doi: 10.1016/j.cclet.2025.111026
S. Ohtani, S. Akine, K. Kato, et al., J. Am. Chem. Soc. 146 (2024) 4695–4703. doi: 10.1021/jacs.3c12093
S. Ohtani, K. Nakaguchi, K. Kato, et al., Chem. Asian J. 19 (2024) e202400106. doi: 10.1002/asia.202400106
W. Xue, Y. Liu, H. Li, Y. Yin, et al., Org. Lett. 27 (2025) 528–532. doi: 10.1021/acs.orglett.4c04607
S.I. Etkind, S. Ichii, N.A. Romero, et al., Synlett 33 (2022) 1532–1538. doi: 10.1055/s-0040-1719932
N. Nikishkin, J. Cějka, V. Eigner, et al., J. Org. Chem. 88 (2023) 12357–12366. doi: 10.1021/acs.joc.3c01093
M.P. Sonawane, J. Jacobs, J. Thomas, et al., Chem. Commun. 49 (2013) 6310–6312. doi: 10.1039/c3cc42984a
M.S. Collins, M.E. Carnes, B.P. Nell, et al., Nat. Commun. 7 (2016) 11052. doi: 10.1038/ncomms11052
N. Morohashi, F. Narumi, N. Iki, et al., Chem. Rev. 106 (2006), 5291–5316. doi: 10.1021/cr050565j
L.P. Yang, X. Wang, H. Yao, et al., Acc. Chem. Res. 53 (2020) 198–208. doi: 10.1021/acs.accounts.9b00415
J. Thomas, A.S. Gusak, W. Dehaen, Homoheteracalix[n]arenes (X= S, Se, N), in: P. Neri, J.L. Sessler, M.X. Wang (Eds.), Calixarenes and Beyond, Springer International Publishing, Cham, 2016, pp. 421–444.
E. Nieland, J. Voss, A. Mix, et al., Angew. Chem. Int. Ed. 61 (2022) e202212745. doi: 10.1002/anie.202212745
Y. Lei, Z. Li, G. Wu, et al., Nat. Commun. 13 (2022) 3557. doi: 10.1038/s41467-022-31289-1
B.P. Benke, T. Kirschbaum, J. Graf, et al. Nat. Chem. 15 (2023) 413. doi: 10.1038/s41557-022-01094-w
X. Kong, Z. Wu, M. Strømme, et al., J. Am. Chem. Soc., 146 (2024) 742–751. doi: 10.1021/jacs.3c10691
C.D. Meyer, C.S. Joiner, J.F. Stoddart, Chem. Soc. Rev. 36 (2007) 1705–1723. doi: 10.1039/b513441m
Y. Ding, L.O. Alimi, J. Du, et al., Chem. Sci. 13 (2022) 3244–3248. doi: 10.1039/d2sc00207h
N.E. Borisova, M.D. Reshetova, Y.A. Ustynyuk, Chem. Rev. 107 (2007) 46–79. doi: 10.1021/cr0683616
Z. He, G. Ye, W. Jiang, Chem. Eur. J. 21 (2015) 3005–3012. doi: 10.1002/chem.201405912
Z. Yang, F. Esteve, C. Antheaume, et al., J. Am. Chem. Soc. 146 (2024) 15438–15445. doi: 10.1021/jacs.4c03698
Y. Pang, M. Wang, N. -H. Yang, et al., Chin. Chem. Lett. 35 (2024) 10957.
K. Xu, B. Li, S. Yao, et al., Angew. Chem. Int. Ed. 61 (2022) e202203016. doi: 10.1002/anie.202203016
J. Jazwinski, J. -M. Lehn, R. Méric et al., Tetrahedron. Lett. 28 (1987) 3489–3492. doi: 10.1016/S0040-4039(00)96334-2
J. Lisowski, Molecules 27 (2022) 4097. doi: 10.3390/molecules27134097
T. Ogoshi, T. Aoki, K. Kitajima, et al., J. Org. Chem. 76 (2011) 328–331. doi: 10.1021/jo1020823
J. Hu, S. Bleus, L. Achten, et al., J. Membr. Sci., 692 (2024) 122282.
P.T. Corbett, J. Leclaire, L. Vial, et al., Chem. Rev. 106 (2006) 3652–3711. doi: 10.1021/cr020452p
X. Shu, S. Chen, J. Li, et al., Chem. Commun. 48 (2012) 2967–2969. doi: 10.1039/c2cc00153e
S. Dong, B. Zheng, Y. Yao, et al., Adv. Mater. 25 (2013) 6864–6867. doi: 10.1002/adma.201303652
M. -S. Yuan, H. Chen, X. Du, et al., Chem. Commun. 71 (2015) 16361–16364.
Y. Yang, T.K. Ronson, J. Zheng, et al., Nat. Commun. 12 (2021) 4079.
T.K. Ronson, J.P. Carpenter, J.R. Nitschke, Chem 8 (2022) 557–568.

Scheme 1 Three-step synthesis of pillarrhombimine PIm, proton allocation based on 1H and NOESY NMR, and structures of nitrile guests G1-G5.

Figure 1 Stick representation of (a) the asymmetric unit of PIm, (b) side-view of the PIm solid-state dimers and (c) packing showing 1D polymeric motifs along the a-axis. Hydrogen atoms are omitted for clarity.

Figure 2 Rotational free-energy of permethoxypillar[5]arene (P5; black circles) and pillarrhombimine (PIm; blue triangles) at the B3LYP/6–311G(2d,2p) level of theory in chloroform.

Figure 3 DFT-optimized host-guest complexes of (a) G1@PIm, (b) G3@PIm, (c) G4@PIm, (d) G5@PIm at the B97D3/SVP/SVPFIT level of theory in chloroform.

Figure 4 Partial 1H NMR spectra (CDCl3, 400 MHz, 298 K, 5.0 × 10–3 mol/L) of PIm (top), G3 (middle) and 1:1 G3@PIm (bottom).

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:

 下载:
下载: