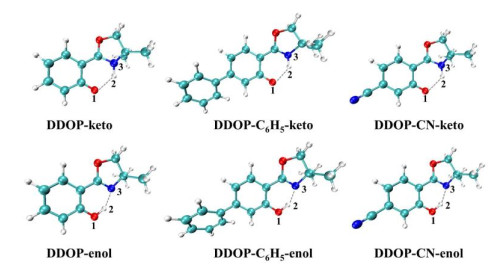
Figure 1.
Optimized structures of the enol forms in the S0 state and keto forms in the S1 state for DDOP, DDOP-C6H5, and DDOP-CN
Computational Insights into the Excited State Intramolecular Proton Transfer Reactions in Ortho-hydroxylated Oxazolines
Gai-Mei LIU , Wei-Jia MA , Yan WANG , Yan YANG , Xin-Jian SONG

Hydrogen bond, as one of most fundamental weak interactions, has been an important research subjects for a long time due to its pervasiveness in biology, physics and chemistry fields[1-9]. It plays a critical role in the stabilization of polypeptides and proteins as well as the accumulation of crystals. Since the dynamics of excited hydrogen bonds were proposed, excited state hydrogen bond was widely found in many photophysical and photochemical processes, such as intramolecular charge transfer (ICT)[10, 11], photoinduced electron transfer (PET)[12, 13], fluorescence resonance energy transfer (FRET)[14], fluorescence quenching (FQ)[15, 16], and excited state intramolecular proton transfer (ESIPT)[17]. ESIPT reaction with hydrogen bonding should be one of the most important dynamic processes. Commonly, the molecules with ESIPT properties involve a heterocyclic ring which possesses proton donor (-OH or -NH2) and acceptor (-C=O or -N=) bridged by intramolecular hydrogen bond. In excited states, the proton would transfer from donor to acceptor rapidly. Previous work has shown that the ESIPT reaction was a process of four-level enol-keto-phototautomerism cycle[18-22], in which, dual fluorescence and a large stokes shift between absorption and tautomer emission could be observed[23, 24]. These remarkable natures make ESIPT molecules widely developed in a variety of applications, fluorescence sensors, laser dyes, UV-absorbers, molecular switches, fluorescent probes, bioimaging and OLEDs[25-31].
ESIPT compounds include the derivatives of salicylate and aniline salicylate, flavonoids, benzoazoles and chalcolones[32]. Their photophysical properties could be modulated via introducing electron withdrawing and donating substituents, replacing heteroatom or changing the conjugation of structures[33]. Up to now, the effects of substituents have received intense attention both experimentally and theoretically. By introducing different substituents into the benzothiazolyl ring, Li et al. synthesized a series of novel 2-(2-hydroxyphenyl)benzothiazole (HBT) derivatives[34]. The emission intensity of these derivatives decreases with the enhancement of electron-withdrawing ability of the substituents and further demonstrated that electron-withdrawing substituents are not beneficial to the ESIPT reactions. In the experiment, Araki's team observed the blue- and red-shift of fluorescence emission peaks of 1-(2΄-hydroxyphenyl)-1H-imidazo[4, 5-c]pyridine (HPIP) derivatives would be tuned by introducing electron withdrawing and electron donating groups into the part of proton donor, respectively[35]. However, the introduction of such groups into the part of proton accepter led to opposite results. Tong and co-workers investigated the asymmetric substitution effect on the optical properties of keto-salicylaldehyde azine (KSA), which is valuable to design and develop novel KSA fluorescent probes[36]. Very recently, Nachtsheim and co-workers have developed an efficient method to synthesize oxazoline-directed ortho C(sp2)–H hydroxylation using molecular oxygen or air as green oxidants. The emission properties of selected phenols were investigated in dichloromethane, showing almost exclusive ESIPT keto-emission and exhibiting large Stokes shifts up to 12, 000 cm-1. Depending on the substitution pattern and the π-extension of luminophore, the emission wavelengths range from blue to green and red[37]. However, the deeper investigations into luminescent properties and the detailed ESIPT dynamical behaviors of ortho-hydroxylated oxazolines are deficient. In this study, the ESIPT reactions and spectral properties of three typical ortho-hydroxylated oxazolines, 2-(4, 4-dimethyl-4, 5-dihydrooxazol-2-yl)-phenol (DDOP), 4-(4, 4-dimethyl-4, 5-dihydrooxazol-2-yl)-[1, 1΄-biphenyl]-3-ol (DDOP-C6H5) and 4-(4, 4-dimethyl-4, 5-dihydrooxazol-2-yl)-3-hydroxy-ben-zoni-trile (DDOP-CN) (Fig. 1) were systemically investigated in dichloromethane solvent by using DFT and TD-DFT methods. The geometries for different electronic states were optimized and their bond lengths, bond angles and infrared vibrational spectra associated with the hydrogen bonds were analyzed. The frontier molecular orbitals (FMOs) and reduced density gradient (RDG) function were also discussed. To further reveal the ESIPT reactions, we constructed the S0 and S1 state potential energy curves.
In this work, all the quantum-chemical computations about the electronic structures of the ground states (S0) and the first excited states (S1) were obtained on the basis of DFT and TDDFT methods with B3LYP functional[38] in combination with 6-31+G(d) basis set by Gaussian 09 programs[39]. All the compounds were optimized with no constraints, and the most stable structures are given without imaginary frequencies. The absorption and emission properties were calculated based on the optimized S0 and S1 structures. Simultaneously, the experimental environment was simulated in view of the polarizable continuum model (PCM) incur-porating dichloromethane as the solvent[40-42]. In order to precisely clarify the ESIPT mechanism, the potential energy curves (PECs) in the S0 and S1 states were constructed by scanning the O(1)–H(2) bond length for a fixed step size from 0.8 to 2.2 Å at a step of 0.1 Å. In addition, the reduced density gradient (RDG) analysis was obtained by Multiwfn[43] and VMD[44] softwares.
All geometry structures of the S0 and S1 states of three ortho-hydroxylated oxazolines (DDOP, DDOP-C6H5, and DDOP-CN) are optimized in dichloromethane solvent based on B3LYP/6-31+G(d) and TD-B3LYP/6-31+G(d) levels. The optimized enol forms in the S0 states and keto forms in the S1 states of DDOP, DDOP-C6H5 and DDOP-CN are shown in Fig. 1, and the major bond lengths and bond angles of each structure are provided in Table 1. The calculated results show that there are two stable forms (enol and keto) for all compounds in the S0 state, but no stable DDOP-enol and DDOP-CN-enol forms in the S1 states can be optimized. In other words, when we optimized the DDOP-enol(S1) and DDOP-CN-enol(S1) forms, the stable states turn out to be DDOP-keto (S1) and DDOP-CN-keto (S1) forms. It shows that the ESIPT reactions of DDOP and DDOP-CN molecules may be a non-barrier process, which will be discussed in detail in the section of potential energy curves. However, when introducing an electron donating group (-C6H5) at the para position of the phenyl ring, the corresponding molecule is DDOP-C6H5. There are two stable forms (enol and keto) in the S1 states, which would lead to an energy barrier in the ESIPT reaction. As a result, the ESIPT reaction of DDOP-C6H5 is more difficult than that of DDOP and DDOP-CN to some extent.
 DownLoad:
CSV
DownLoad:
CSV
| DDOP-enol | DDOP-keto | DDOP-C6H5-enol | DDOP-C6H5-keto | DDOP-CN-enol | DDOP-CN-keto | |||||||
| State | S0 | S1 | S0 | S1 | S0 | S1 | S0 | S1 | S0 | S1 | S0 | S1 |
| O(1)−H(2) | 1.000 | - | 1.820 | 2.018 | 1.000 | 1.010 | 1.819 | 2.007 | 1.001 | - | 1.826 | 2.011 |
| H(2)−N(3) | 1.750 | - | 1.032 | 1.025 | 1.750 | 1.710 | 1.032 | 1.023 | 1.742 | - | 1.032 | 1.022 |
| O(1)−H(2)···N(3) | 146.7 | - | 131.5 | 127.0 | 146.8 | 148.8 | 131.5 | 126.6 | 146.5 | - | 131.1 | 125.2 |
In view of the DDOP-C6H5-enol form, it can be seen that DDOP-C6H5 changed from S0(enol) to S1(enol) upon photoexcitation, O(1)−H(2) bond length increased from 1.000 to 1.010 Å, and the H(2)···N(3) bond length reduced significantly from 1.750 to 1.710 Å. In addition, the O(1)–H(2)···N(3) bond angle in the S0 state (146.8°) increased significantly in the S1 state (148.8°). The lengthening of the O(1)–H(2) bond length, the shortening of the H(2)···N(3) bond length and the increase of O(1)–H(2)···N(3) bond angle suggest that the hydrogen-bond O(1)–H(2)···N(3) is reinforced in the S1 state, which can facilitate the ESIPT reaction. As to the keto forms, in the S1 state, the O(1)···H(2) bond lengths of DDOP, DDOP-C6H5, and DDOP-CN are 2.018, 2.007 and 2.011 Å, and the H(2)–N(3) bond lengths are 1.025, 1.025 and 1.022 Å, respectively, which confirm that the H(2) atoms have migrated from O(1) atoms to N(3) atoms and form new covalent bonds with N(3) atoms. In addition, the O(1)···H(2) bond lengths of DDOP, DDOP-C6H5, and DDOP-CN are decreased to 1.820, 1.819 and 1.826 Å in the S0 states. And the H(2)–N(3) bond lengths are increased to 1.320 Å in the S0 states. Meanwhile, the bond angles O(1)···H(2)–N(3) are enlarged from 127.0°, 126.6° and 125.2° in the S1 states to 131.5°, 131.5° and 131.1° in the S1 states. It can be concluded that the hydrogen bonds O(1)···H(2)–N(3) are more stable in the S0 states. That is, the keto forms of the S1 states are likely to undergo radiative transition to the S0 states, forming stable intramolecular hydrogen bond O(1)···H(2)–N(3) after the ESIPT process.
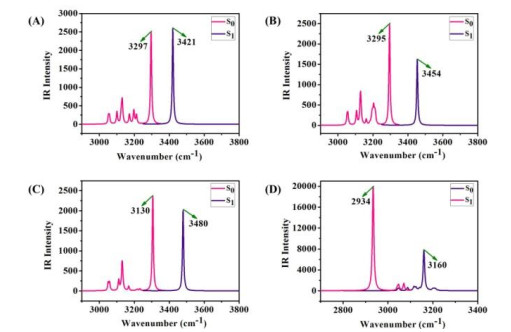
As is well known, an effective signature for the evidence of excited state hydrogen bond strengthening or weakening can be estimated by the peak red-shift or blue-shift of the stretching vibrations of O–H moiety involved in hydrogen bond[45-48]. In this work, the infrared spectra were performed in both S0 and S1 states, and the infrared vibrational spectra linking the hydrogen bond in dichloromethane solvent are shown in Fig. 2. As can be seen, the S1 states vibration frequencies of H(2)–N(3) group in DDOP-keto, DDOP-C6H5-keto, and DDOP-CN-keto are 3421, 3454, and 3480 cm-1, decreased to 3297, 3295, and 3130 cm-1 in the S0 states, respectively. The 124, 159, and 350 cm-1 blue-shifts of H(2)–N(3) stretching frequency obviously reveal the O(1)–H(2)···N(3) hydrogen bonds of these three compounds are strengthened in the S0 states. For DDOP-C6H5-enol, the computed O(1)–H(2) stretching vibrational frequency is located at 3160 cm-1 in the S0 state, whereas 2934 cm-1 in the S1 state. A significant red-shift of 226 cm-1 for the O(1)–H(2) stretching band indicates that the O(1)–H(2)···N(3) hydrogen bond is strengthened in the S1 state. Therefore, the ESIPT reaction of DDOP-C6H5 might be promoted by the strengthened hydrogen bond, which is in agreement with the result based on analyzing bond lengths and bond angles.
The calculated Stokes shifts, absorption and emission peaks, oscillator strengths (f), and the corresponding compositions based TD-B3LYP/6-31+G(d) level are reported in Table 2, together with the available experimental values[37]. It should be noted that the calculated absorption peak of DDOP, DDOP-C6H5, and DDOP-CN are 287, 306 and 311 nm, respectively, which are in good agreements with the corresponding experimental data (303, 316 and 326 nm)[37]. The calculated fluorescence emission peaks based on the optimized S1 states of DDOP-keto, DDOP-C6H5-keto, and DDOP-CN-keto are located at 450, 447 and 446 nm, which also match well with the experimental ESIPT-based emission values (470, 472 and 471 nm). In addition, all three compounds show large Stokes shifts, the calculated Stokes shifts of DDOP, DDOP-C6H5, and DDOP-CN are 12621 cm-1, 10308, and 9733, which are well consistent with the experimental values (11730, 10460, and 9440 cm-1). That is to say, the absence of the emission of the enol form would confirm that the ESIPT reactions are instantaneously in the S1 states. In a word, the experimental absorption and emission spectra are well reproduced based on our calculated results, which demonstrate that the theoretical level is reasonable and effective.
DownLoad:
CSV
| Compound | Absorption | Emission | Stokes shift (cm-1) |
Stokes shift exp(cm-1) |
|||||||
| Composition (CI%) | λ(nm) | f | λexp(nm) | Composition (CI%) | λ(nm) | f | λexp(nm) | ||||
| DDOP | HOMO→LUMO(92%) | 287 | 0.1567 | 303 | HOMO→LUMO(97%) | 450 | 0.1328 | 470 | 12621 | 11730 | |
| DDOP-C6H5 | HOMO→LUMO(90%) | 306 | 0.4672 | 316 | HOMO→LUMO(98%) | 447 | 0.1345 | 472 | 10308 | 10460 | |
| DDOP-CN | HOMO→LUMO(93%) | 311 | 0.2072 | 326 | HOMO→LUMO(97%) | 446 | 0.1598 | 471 | 9733 | 9440 | |
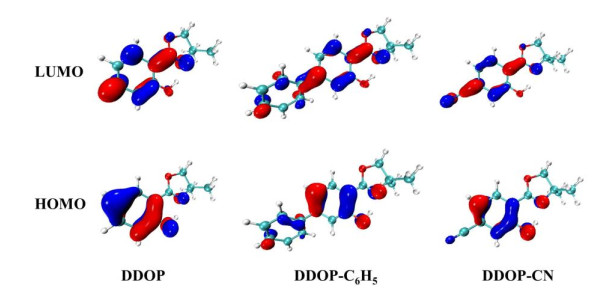
In addition, it is well known that the nature of the excited state can be directly exhibited by analyzing the frontier molecular orbitals (FMOs). The highest occupied orbital (HOMO) and the lowest unoccupied orbital (LUMO) are displayed in Fig. 3. The S0→S1 transition is mainly relative to the HOMO→LUMO for DDOP, DDOP-C6H5, and DDOP-CN molecules, in which their orbital transition contribution rates are 92%, 90% and 93%, as shown in Table 2. It can be distinctly found that the excitation processes have significant characteristic π→π* transitions from the HOMO to the LUMO for these compounds. As shown in Fig. 3, the electron density distributions of HOMO and LUMO are different. Herein, we mainly focus on the differences about the moiety involved in intramolecular H-bond O(1)–H(2)···N(3). The HOMO→LUMO transition make the electron density of O(1) atoms decrease and the electron densities of N(3) atom increase. As a result, the intramolecular hydrogen bond O(1)–H(2)···N(3) is strengthened, which may further trigger the proton transfer.
To further elucidate the mechanism of ESIPT reactions in the ortho-hydroxylated oxazolines, the S0 and S1 states potential energy curves of DDOP, DDOP-C6H5 and DDOP-CN have been scanned along the proton transfer pathways based on constrained optimizations at the fixed O(1)–H(2) distance from 0.8 to 2.2 Å in a step of 0.1 Å, and the potential energy curves are shown in Fig. 4. The potential energy curves show two minimum energy points in the S0 state, and the potential barriers are 6.26, 6.24 and 5.58 kcal·mol-1 for DDOP, DDOP-C6H5 and DDOP-CN, respectively. It indicates that the stable enol forms can barely convert into the keto ones because of their high energy barriers for these three compounds. In comparison, the energy barriers for the RGSIPT processes are 3.46, 3.51 and 3.91 kcal·mol-1 for DDOP, DDOP-C6H5 and DDOP-CN, respectively, which are significantly lower than that of the GSIPT processes. That is to say, the RGSIPT processes are easier to occur than that of the GSIPT processes to some extent.
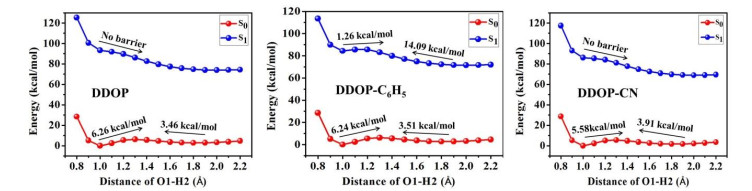
For the S1 states, there is only minimum energy point for DDOP and DDOP-CN, and the ESIPT reactions are assumed to be a barrierless process, which are in accordance with the optimized geometries and explain why the optimized geometries of DDOP-enol (S1) and DDOP-CN-enol (S1) are not obtained. It means that the proton transfer processes are spontaneously in the S1 state. In contrast to DDOP and DDOP-CN, there are two minimum energy points and a small barrier of 1.26 kcal·mol-1 along the ESIPT process for DDOP-C6H5, indicating that the DDOP-C6H5-enol can be easily isomerized into DDOP-C6H5-keto (minimum energy point) by crossing a low barrier in the S1 state. Hence, it could conceivably be concluded that the donating group (-C6H5) at the para position of the phenyl ring hinders the ESIPT reactions. Furthermore, the reverse proton transfer barrier of DDOP-C6H5 is relatively higher (14.09 kcal·mol-1), and the DDOP-C6H5-keto can barely convert into DDOP-C6H5-enol. As a result, there is no enol form emission due to the high reverse proton transfer barrier and the ultrafast ESIPT process following the excitation, which is in accordance with the experimental emission spectra[37].
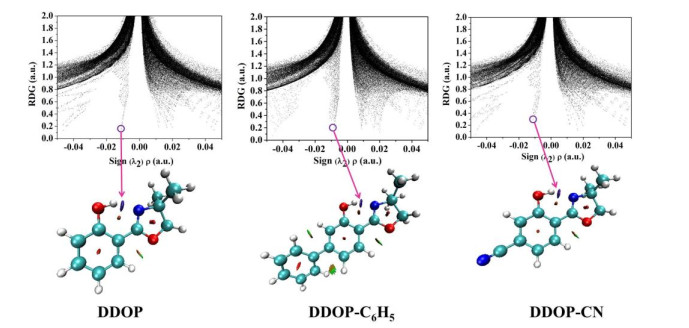
To visualize the hydrogen bonding interactions in the real space, we calculated the reduced density gradient (RDG) scatter plots and the corresponding isosurfaces in the S0 state. As reported by previous work[49], the positive values of sign(λ2)ρ, the values of sign(λ2)ρ close to zero and the negative sign(λ2)ρ represent the steric effect, Van der Waals (VDW) interactions and hydrogen bonding interactions, respectively. As shown in Fig. 5, the spikes locates around –0.01 a.u. in the S0 state. This phenomenon once again illustrates the intramolecular hydrogen bond O(1)–H(2)···N(3) for DDOP and its derivatives.
In this present work, DFT and TD-DFT calculations with B3LYP functional in combination with the 6-31+G(d) basis set are performed to explore the ESIPT mechanisms of three ortho-hydroxylated oxazolines (DDOP, DDOP-C6H5, and DDOP-CN), and their absorption and fluorescence spectra are simulated and their potential energy curves involving the S0 and S1 states are constructed. Via analyzing the bond lengths, bond angles, and infrared vibrational spectra of these three stable structures, we confirm that the intramolecular hydrogen bond O(1)−H(2)···N(3) should be strengthened in the S1 state. All three compounds show large Stokes shifts of ~10000 cm-1 due to the absence of the emission of enol form. Our calculated results reproduce well the experimental absorption and emission spectra and the first electronic transitions of all three compounds have significant characteristic π→π* nature. The HOMO→LUMO transition results in the redistribution of charge and would facilitate the ESIPT reaction. In addition, the constructed potential energy curves of both S0 and S1 states further confirm that the proton transfer reactions can take place in the S1 state easier than in the S0 state. More importantly, the S1 proton transfer potential energy curves of DDOP and DDOP-CN have been determined to be barrierless, indicating a fast dynamics mechanism, but there is a small barrier for DDOP-C6H5, which further verifies that the electron donating is unfavorable to the ESIPT reactions of ortho-hydroxylated oxazolines.
Chen, M. J.; Runge, T.; Wang, L. L.; Li, R.; Feng, J.; Shu, X. L.; Shi, Q. S. Hydrogen bonding impact on chitosan plasticization. Carbohyd. Polym. 2018, 200, 115−121. doi: 10.1016/j.carbpol.2018.07.062
Zuo, H. Y.; Christopher, J. R.; Wong, T. H. F.; Tong, K. Y.; Chan, J.; Au-Yeung, H. Y. Activity-based sensing of ascorbate by using copper-mediated oxidative bond cleavage. Chem. Eur. J. 2020, 26, 1−8. doi: 10.1002/chem.201905547
Dalchand, N.; Cui, Q.; Geiger, F. M. Electrostatics, hydrogen bonding, and molecular structure at polycation and peptide: lipid membrane interfaces. ACS Appl. Mater. Inter. 2020, 12, 2149−2158.
Sessler, C. D.; Rahm, M.; Becker, S.; Goldberg, J. M.; Wang, F.; Lippard, S. J. CF2H, a hydrogen bond donor. J. Am. Chem. Soc. 2017, 139, 9325−9332. doi: 10.1021/jacs.7b04457
Yang, Z. G.; Cao, J. F.; He, Y. X.; Yang, J. H.; Kim, T.; Peng, X. J.; Kim, J. S. Macro-/micro-environment-sensitive chemosensing and biological imaging. Chem. Soc. Rev. 2014, 43, 4563−4601. doi: 10.1039/C4CS00051J
Brovarets', O. O.; Yurenko, Y. P.; Hovorun, D. M. Intermolecular СН···О/N Н-bonds in the biologically important pairs of natural nucleobases: a thorough quantum-chemical study. J. Biomol. Struct. Dyn. 2014, 32, 993−1022. doi: 10.1080/07391102.2013.799439
Liang, S. Z.; Hammond, G. B.; Xu, B. Hydrogen bonding: regulator for nucleophilic fluorination. Chem. Eur. J. 2017, 23, 17850−17861. doi: 10.1002/chem.201702664
Crabtree, R. H. Hypervalency, secondary bonding and hydrogen bonding: siblings under the skin. Chem. Soc. Rev. 2017, 46, 1720−1729. doi: 10.1039/C6CS00688D
Robertson, C. C.; Wright, J. S.; Carrington, E. J.; Perutz, R. N.; Hunter, C. A.; Brammer, L. Hydrogen bonding vs. halogen bonding: the solvent decides. Chem. Sci. 2017, 8, 5392−5398. doi: 10.1039/C7SC01801K
Wen, Z. C.; Jiang, Y. B. Ratiometric dual fluorescent receptor for anions under intramolecular charge transfer mechanism. J. Cheminform. 2005, 36, 11109−11115.
Kumari, N.; Jha, S.; Bhattacharya, S. Colorimetric probes on anthraimidazolediones for selective sensing of fluoride and cyanide ion via intramolecular charge transfer. J. Organomet. Chem. 2011, 76, 8215−8222.
Khrenova, M. G.; Nemukhin, A. V.; Domratcheva, T. Photoinduced electron transfer facilitates tautomerization of the conserved signaling glutamine side chain in BLUF protein light sensors. J. Phys. Chem. B 2013, 117, 2369−2377. doi: 10.1021/jp312775x
Hankache, J.; Hanss, D.; Wenger, O. S. Hydrogen-bond strengthening upon photoinduced electron transfer in ruthenium-anthraquinone dyads interacting with hexafluoroisopropanol or water. J. Phys. Chem. A 2012, 116, 3347−3358. doi: 10.1021/jp300090n
Xu, W. J.; Liu, S. J.; Sun, H. B.; Zhao, X. Y.; Zhao, Q.; Sun, S.; Cheng, S.; Ma, T. C.; Zhou, L. X.; Huang, W. RET-based probe for fluoride based on a phosphorescent iridium(III) complex containing triarylboron groups. J. Mater. Chem. 2011, 21, 7572−7581. doi: 10.1039/c1jm00071c
Barman, N.; Singha, D.; Sahu, K. Fluorescence quenching of hydrogen-bonded coumarin 102-phenol complex: effect of excited-state hydrogen bonding strength. J. Phys. Chem. A 2013, 117, 3945−3953. doi: 10.1021/jp4019298
Chang, D. H.; Ou, C. L.; Hsu, H. Y.; Huang, G. J.; Kao, C. Y.; Liu, Y. H.; Peng, S. M.; Diau, E. W. G.; Yang, J. S. Cooperativity and site-selectivity of intramolecular hydrogen bonds on the fluorescence quenching of modified GFP chromophores. J. Org. Chem. 2015, 80, 12431−12443. doi: 10.1021/acs.joc.5b02303
Cao, H.; Liu, G. M.; Cai, J.; Wang, Y. Excited-state intramolecular proton transfer mechanisms of thiazole-based chemosensor: a TD-DFT study. Chin. J. Struct. Chem. 2020, 11, 1993−1940.
Yang, D. P.; Yang, G.; Jia, M.; Song, X. Y.; Zhang, Q. L. Comparing the substituent effects about ESIPT process for HBO derivatives. Comput. Theor. Chem. 2018, 1131, 51−56. doi: 10.1016/j.comptc.2018.03.016
Li, Y. Q.; Ma, Y. Z.; Yang, Y. F.; Shi, W.; Lan, R. F.; Guo, Q. Effects of different substituents of methyl 5-R-salicylates on the excited state intramolecular proton transfer process. Phys. Chem. Chem. Phys. 2018, 20, 4208−4215. doi: 10.1039/C7CP06987A
Berbigier, J. F.; Duarte, L. G. T. A.; Zawacki, M. F.; Araújo, B. B.; Santos, C. M.; Atvars, T. D. Z.; Gonçalves, P. F. B.; Petzhold, C. L.; Rodembusch, F. S. ATRP initiators based on proton transfer benzazole dyes: solid state photoactive polymer with very large stokes shift. ACS Appl. Polym. Mater. 2020, 2, 1406−1416. doi: 10.1021/acsapm.0c00102
Padalkar, V. S.; Seki, S. Excited-state intramolecular proton-transfer (ESIPT)-inspired solid state emitters. Chem. Soc. Rev. 2015, 45, 169−202.
Kwon, J. E.; Park, S. Y. Advanced organic optoelectronic materials: harnessing excited-state intramolecular proton transfer (ESIPT) process. Adv. Mater. 2011, 23, 3615−3642. doi: 10.1002/adma.201102046
Sedgwick, A. C.; Wu, L. L.; Han, H. H.; Bull, S. D.; He, X. P.; James, T. D.; Sessler, J. L.; Tang, B. Z.; Tian, H.; Yoon, J. Excited-state intramolecular proton-transfer (ESIPT) based fluorescence sensors and imaging agents. Chem. Soc. Rev. 2018, 47, 8842−8880. doi: 10.1039/C8CS00185E
Zhang, Q. L.; Zhao, Z. J.; Cheng, S. B.; Yang, G.; Zhang, T. J.; Jia, M.; Song, X. Y. A theoretical investigation on the excited state intramolecular single or double proton transfer mechanism of a salicyladazine system. J. Chin. Chem. Soc. 2019, 66, 1416−1421. doi: 10.1002/jccs.201800490
Zhang, Y. W.; Sun, Q. K.; Li, Z. P.; Zhi, Y. F.; Li, H.; Li, Z. P.; Xia, H.; Liu, X. M. Light-emitting conjugated microporous polymers based on an excited-state intramolecular proton transfer strategy and selective switch-off sensing of anions. Mater. Chem. Front. 2020, 4, 3040−3046. doi: 10.1039/D0QM00384K
Sugiyama, K.; Tsuchiya, T.; Kikuchi, A.; Yagi, M. Optical and electron paramagnetic resonance studies of the excited triplet states of UV-B absorbers: 2-ethylhexyl salicylate and homomenthyl salicylate. Photochem. Photobiol. Sci. 2015, 14, 1651−1659. doi: 10.1039/c5pp00138b
Kanamori, D.; Okamura, T. A.; Yamamoto, H.; Ueyama, N. Linear-to-turn conformational switching induced by deprotonation of unsymmetrically linked phenolic oligoamides. Angew. Chem. Int. Ed. 2005, 44, 969−972. doi: 10.1002/anie.200461842
Yan, C. C.; Wang, X. D.; Liao, L. S. Organic lasers harnessing excited state intramolecular proton transfer process. ACS Photonics. 2020, 7, 1355−1366. doi: 10.1021/acsphotonics.0c00407
Zhao, Y.; Ding, Y.; Yang, Y.; Shi, W.; Li, Y. Fluorescence deactivation mechanism for a new probe detecting phosgene based on ESIPT and TICT. Org. Chem. Front. 2019, 6, 597−602. doi: 10.1039/C8QO01320A
Kaur, I.; Sharma, V.; Mobin, S. M.; Khajuria, A.; Ohri, P.; Kaur, P.; Singh, K. Aggregation tailored emission of a benzothiazole based derivative: photostable turn on bioimaging. RSC Adv. 2019, 9, 39970−39975. doi: 10.1039/C9RA08149F
Duarte, L. T.; Germino, J. C.; Berbigier, J. F.; Barboza, C. A.; Faleiros, M. M.; Simoni, D. A.; Galante, M. T.; Holanda, M. S.; Rodembusch, F. S.; Atvars, T. D. Z. White-light generation from all-solution-processed OLEDs using a benzothiazole-salophen derivative reactive to the ESIPT process. Phys. Chem. Chem. Phys. 2019, 21, 1172−1182. doi: 10.1039/C8CP06485G
Watwiangkham, A.; Roongcharoen, T.; Kungwan, N. Effect of nitrogen substitution and conjugation on photophysical properties and excited state intramolecular proton transfer reactions of methyl salicylate derivatives. J. Photoch. Photobio. A Chem. 2019, 389, 112267−11.
Berbigier, J. F.; Duarte, L. T.; Zawacki, M.; Araújo, B.; Santos, C.; Atvars, T. Z.; Goncalves, P. F. B.; Petzhold, C. L.; Rodembusch, F. S. ATRP initiators based on proton transfer benzazole dyes: solid state photoactive polymer with very large stokes shift. ACS Appl. Polym. Mater. 2020, 2, 1406−1416. doi: 10.1021/acsapm.0c00102
Wang, R. J.; Liu, D.; Xu, K.; Li, J. Y. Substituent and solvent effects on excited state intramolecular proton transfer in novel 2-(2-hydroxyphenyl) benzothiazole derivatives. J. Photochem. Photobio. A Chem. 2009, 205, 61−69. doi: 10.1016/j.jphotochem.2009.03.025
Mutai, T.; Sawatani, H.; Shida, T.; Shono, H.; Araki, K. Tuning of excited-state intramolecular proton transfer (ESIPT) fluorescence of imidazo[1, 2 a]pyridine in rigid matrices by substitution effect. J. Org. Chem. 2013, 78, 2482−2489. doi: 10.1021/jo302711t
Tong, J. L.; Zhang, K. X.; Wang, J.; Li, H.; Zhou, F.; Wang, Z. M.; Zhang, X. J.; Tang, B. Z. Keto-salicylaldehyde azine: asymmetric substituent effect on their optical properties via electron-donating group insertion. J. Mater. Chem. C 2020, 8, 996−1001. doi: 10.1039/C9TC05822B
Göbel, D.; Clamor, N.; Lork, E.; Nachtsheim, B. J. Aerobic C(sp2)-H hydroxylations of 2-aryloxazolines: fast access to excited-state intramolecular proton transfer (ESIPT)-based luminophores. Org. Lett. 2019, 21, 5373−5377. doi: 10.1021/acs.orglett.9b01350
Becke, A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648−5652. doi: 10.1063/1.464913
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenberg, J. L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery Jr., J. A.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, J. M.; Klene, M.; Knox, J. E.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Dapprich, S.; Daniels, A. D.; Farkas, O.; Foresman, J. B.; Ortiz, J. V.; Cioslowski, J.; Fox, D. J. Gaussian 09, Revision B. 01, Gaussian, Inc., Wallingford CT 2009.
Mennucci, B.; Cancès, E.; Tomasi, J. Evaluation of solvent effects in isotropic and anisotropic dielectrics and in ionic solutions with a unified integral equation method: theoretical bases, computational implementation, and numerical applications. J. Phys. Chem. B 1997, 101, 10506−10517. doi: 10.1021/jp971959k
Cammi, R.; Tomasi, J. Remarks on the use of the apparent surface charges (ASC) methods in solvation problems: iterative versus matrix-inversion procedures and the renormalization of the apparent charges. J. Comput. Chem. 1995, 16, 1449−1458. doi: 10.1002/jcc.540161202
Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilizaion of ab initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117−129. doi: 10.1016/0301-0104(81)85090-2
Tian, L.; Chen, F. W. Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580−592. doi: 10.1002/jcc.22885
Humphery, W.; Dalke, A.; Shulten, K. VMD: visual molecular dynamics. J. Mol. Graph. 1996, 14, 33−38. doi: 10.1016/0263-7855(96)00018-5
Zhao, G. J.; Han, K. L. Hydrogen bonding in the electronic excited state. Acc. Chem. Res. 2012, 45, 404−413. doi: 10.1021/ar200135h
Zhao, G. J.; Han, K. L. pH-Controlled twisted intramolecular charge transfer (TICT) excited state via changing the charge transfer direction. Phys. Chem. Chem. Phys. 2010, 12, 8914−8918. doi: 10.1039/b924549a
Zhao, G. J.; Han, K. L. Early time hydrogen-bonding dynamics of photoexcited coumarin 102 in hydrogen-donating solvents: theoretical study. J. Phys. Chem. A 2007, 111, 2469−2474. doi: 10.1021/jp068420j
Zhao, G. J.; Han, K. L. Time-dependent density functional theory study on hydrogen-bonded intramolecular charge-transfer excited state of 4-dimethylamino-benzonitrile in methanol. J. Comput. Chem. 2008, 29, 2010−2017. doi: 10.1002/jcc.20957
Johnson, E. R.; Keinan, S.; Mori-Sanchez, P.; Contreras-Carcis, J.; Cohen, A. J.; Yang, W. Revealing noncovalent interactions. J. Am. Chem. Soc. 2010, 132, 6498−6506. doi: 10.1021/ja100936w

Figure 1 Optimized structures of the enol forms in the S0 state and keto forms in the S1 state for DDOP, DDOP-C6H5, and DDOP-CN

Figure 2 Calculated IR vibrational spectra for H(2)−N(3) stretching bands of (A) DDOP-keto, (B) DDOP-C6H5-keto, (C) DDOP-CN-keto and for O(1)−H(2) vibrational mode of (D) DDOP-C6H5-enol

Figure 3 HOMO and LUMO of DDOP, DDOP-C6H5, and DDOP-CN based on the calculated level of TD-DFT/B3LYP/6-31+g(d)

Figure 4 Potential energy curves of the S0 and S1 states for DDOP, DDOP-C6H5, and DDOP-CN along with the proton transfer coordinate

Figure 5 RDG scatter plots and isosurfaces for DDOP, DDOP-C6H5, and DDOP-CN in S0 state
Table 1. Calculated Geometric Parameters (Bond Lengths in Å and Bond Angles in °) for DDOP, DDOP-C6H5, and DDOP-CN in S0 and S1 States Based on the DFT and TD-DFT Methods, respectively
| DDOP-enol | DDOP-keto | DDOP-C6H5-enol | DDOP-C6H5-keto | DDOP-CN-enol | DDOP-CN-keto | |||||||
| State | S0 | S1 | S0 | S1 | S0 | S1 | S0 | S1 | S0 | S1 | S0 | S1 |
| O(1)−H(2) | 1.000 | - | 1.820 | 2.018 | 1.000 | 1.010 | 1.819 | 2.007 | 1.001 | - | 1.826 | 2.011 |
| H(2)−N(3) | 1.750 | - | 1.032 | 1.025 | 1.750 | 1.710 | 1.032 | 1.023 | 1.742 | - | 1.032 | 1.022 |
| O(1)−H(2)···N(3) | 146.7 | - | 131.5 | 127.0 | 146.8 | 148.8 | 131.5 | 126.6 | 146.5 | - | 131.1 | 125.2 |
 下载: 导出CSV
下载: 导出CSV
Table 2. Calculated Absorption and Emission Peaks (nm), Oscillator Strengths (f), and the Corresponding Compositions (CI) for DDOP, DDOP-C6H5, and DDOP-CN in Dichloromethane Solvent, along with the Experimental Data
| Compound | Absorption | Emission | Stokes shift (cm-1) |
Stokes shift exp(cm-1) |
|||||||
| Composition (CI%) | λ(nm) | f | λexp(nm) | Composition (CI%) | λ(nm) | f | λexp(nm) | ||||
| DDOP | HOMO→LUMO(92%) | 287 | 0.1567 | 303 | HOMO→LUMO(97%) | 450 | 0.1328 | 470 | 12621 | 11730 | |
| DDOP-C6H5 | HOMO→LUMO(90%) | 306 | 0.4672 | 316 | HOMO→LUMO(98%) | 447 | 0.1345 | 472 | 10308 | 10460 | |
| DDOP-CN | HOMO→LUMO(93%) | 311 | 0.2072 | 326 | HOMO→LUMO(97%) | 446 | 0.1598 | 471 | 9733 | 9440 | |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载: