Citation:
Shihong Wu, Ronghui Zhou, Hang Zhao, Peng Wu. Sonoafterglow luminescence for in vivo deep-tissue imaging[J]. Chinese Chemical Letters,
2024, 35(10): 110026.
doi:
10.1016/j.cclet.2024.110026
Sonoafterglow luminescence for in vivo deep-tissue imaging
English
Sonoafterglow luminescence for in vivo deep-tissue imaging
Optical imaging is a key technique to study the pathological process and disease diagnosis, but there is still a great challenge to achieve highly sensitive in vivo deep-tissue imaging. Serious interferences from the intrinsic autofluorescence and light scattering and re-absorption are typically observed in biological tissues, which severely impair target signal sensing and acquisition. To address the above issues, using of afterglow luminescence for imaging has been proposed. Due to its long lifetime,the images can be collected after ceasing the excitation, thus could eliminate the autofluorescence. However, the current afterglow luminescence still requires photoexcitation (mainly ultraviolet or visible light), the penetration depth of which is still limited for deep-tissue imaging.
The large tissue penetration of ultrasound (US) has long been recognized, particularly in ultrasonography [1]. Particularly, the cavitation effect (nucleation, growth, and explosion of bubbles under US) of US has been harvested to excite sonosensitizers to produce reactive oxygen species (ROS). It has been demonstrated that the reaction between the US-induced generation of ROS and chemiluminescent substrates could yield persistent luminescence [2], thus is appealing for in vivo deep-tissue imaging.
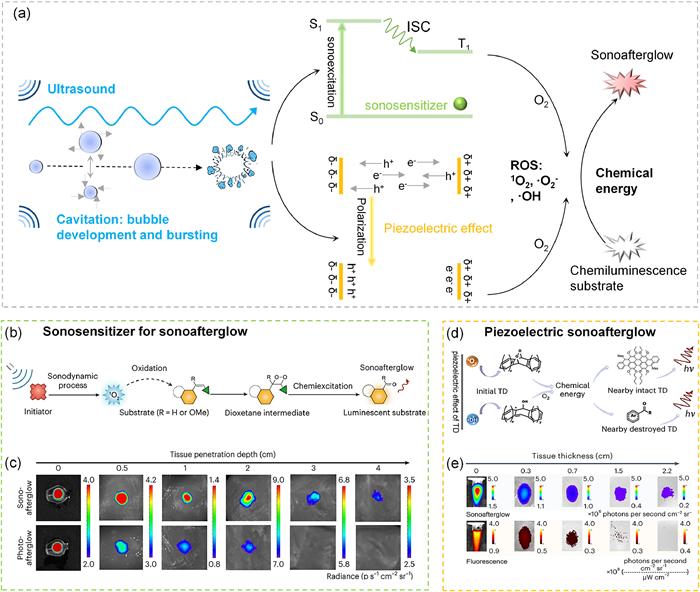
In 2023, Pu and colleagues from Nanyang Technological University first explored the large penetration depth of US and the low background of afterglow luminescence for in vivo imaging (Fig. 1a) [3]. For such goal, they screened a range of organic sonosensitizers as afterglow initiators and polymers or small molecules as afterglow substrates, respectively, to develop sonoafterglow nanoparticles (SNAPs). In this work, the energy flux was as follows: US → sonosenitizer → O2 → 1O2 → substrate → afterglow luminescence. Here, role of US was similar to that of typical photo-excitation. After receiving the US energy, the sonosensitizer experienced a regular photophysical process, namely excitation and then intersystem crossing to produce ROS, which further reacted with sonoafterglow substrates to form self-luminescence dioxetane intermediates to release sonoafterglow luminescence (Fig. 1b). After suitable combinations of sonosensitizers and afterglow substrates, near-infrared afterglow luminescence (780 nm) with a half-life of 110 s was eventually obtained. For in-vivo imaging, the resultant near-infrared (NIR) afterglow luminescence allowed a tissue penetration depth of 4 cm and a high signal-to-background ratio (47.4 times higher than fluorescence, Fig. 1c).
Figure 1
Figure 1.
Sonoafterglow luminescence for deep tissue imaging: (a) ROS generation by sonosensitizers and piezoelectric materials under ultrasonic excitation, and its further two-step energy conversion to obtain sonoafterglow luminescence; (b) sonoafterglow mechanism of SNAPs; (c) sonoafterglow and photoafterglow of NCBS/DPAs SNAP through different thickness tissues; (d) sonoafterglow mechanism of TD NPs; and (e) sonoafterglow and fluorescence of TD NPs through different thickness tissues. (b) and (c) are reproduced with permission [3], Copyright 2023, Springer Nature. (d) and (e) are reproduced with permission [4], Copyright 2024, Springer Nature.
In a very recent work, Zhang and Tan’s group in Hunan University developed a new US-excited afterglow format [4]. Different from Pu’s work, Zhang and Tan utilized the piezoelectric effect to produce ROS for initiating afterglow luminescence (Fig. 1a). Particularly, an organic system, namely trianthracene and its derivatives, was found to be piezoelectrically active. Under ultrasonic agitation, an internal electric field was built. The charge separation eventually lead to the generation of ROS, and thus can be harvested to react with either the chemiluminescent substrate (dioxane) or the adjacent trianthracene derivative-based nanoparticles (TD NPs), and eventually produce afterglow emission (Fig. 1d). After screening, the TD NPs were found to exhibit the highest sonoafterglow luminescence. After ceasing the ultrasonic excitation, the luminescence could be collected over 7 h, with a half-life of 180 s. For in-vivo imaging, a tissue penetration depth of 2.2 cm and a signal-to-noise ratio of 10-fold higher over fluorescence were obtained (Fig. 1e).
To conclude, the above two works clearly demonstrated the potential of US in inducing long-lived afterglow emission. In this manner, deep penetration (US) and high contrast (afterglow emission) in-vivo imaging could be carried out. In the future, the energy collection efficiency of US can be further optimized to increase the luminescence efficiency. Besides, the penetration depth of the emitted light can be further improved, i.e., via NIR-Ⅱ emission. It is obvious that the essence of “ultrasonic → chemical → light” energy conversion is advantageous for deep tissue imaging, and superior and tunable sonoafterglow properties (wavelength, intensity, and half-life) can be expected. Meanwhile, bioluminescent substrates with better biocompatibility and higher luminescence efficiency can be readily introduced for in-situ detection of subtle molecular pathological changes. Overall, these two works opened up a new avenue for the further development of activatable diagnostic probes for real-time, non-invasive, and highly sensitive monitoring of pathological processes and disease diagnosis [5].
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (Nos. 82373394 and 22325403), the Multidisciplinary Research Program of West China Hospital of Stomatology, Sichuan University (No. RD-03-202109), and the Open Research Fund of School of Chemistry and Chemical Engineering, Henan Normal University (No. 2022A02).
Figure 1
Sonoafterglow luminescence for deep tissue imaging: (a) ROS generation by sonosensitizers and piezoelectric materials under ultrasonic excitation, and its further two-step energy conversion to obtain sonoafterglow luminescence; (b) sonoafterglow mechanism of SNAPs; (c) sonoafterglow and photoafterglow of NCBS/DPAs SNAP through different thickness tissues; (d) sonoafterglow mechanism of TD NPs; and (e) sonoafterglow and fluorescence of TD NPs through different thickness tissues. (b) and (c) are reproduced with permission [3], Copyright 2023, Springer Nature. (d) and (e) are reproduced with permission [4], Copyright 2024, Springer Nature.

 DownLoad:
DownLoad:

 下载:
下载:


 下载:
下载:
