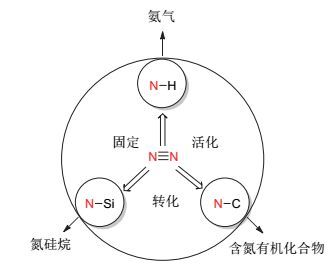
 图 1
氮气转化领域的三个研究方向
Figure 1.
Research directions towards transformations of dinitrogen
图 1
氮气转化领域的三个研究方向
Figure 1.
Research directions towards transformations of dinitrogen
引用本文:
李嘉鹏, 殷剑昊, 俞超, 张文雄, 席振峰. 从氮气直接合成含氮有机化合物[J]. 化学学报,
2017, 75(8): 733-743.
doi:
10.6023/A17040170

Citation: Li Jiapeng, Yin Jianhao, Yu Chao, Zhang Wenxiong, Xi Zhenfeng. Direct Transformation of N2 to N-Containing Organic Compounds[J]. Acta Chimica Sinica, 2017, 75(8): 733-743. doi: 10.6023/A17040170
Citation: Li Jiapeng, Yin Jianhao, Yu Chao, Zhang Wenxiong, Xi Zhenfeng. Direct Transformation of N2 to N-Containing Organic Compounds[J]. Acta Chimica Sinica, 2017, 75(8): 733-743. doi: 10.6023/A17040170
从氮气直接合成含氮有机化合物
摘要:
作为与人类文明和生存密切相关的重大研究方向,温和条件下氮气的活化与转化(固氮)研究在1970~1990年代曾经是国际上备受关注的研究领域.但是,由于该领域极具挑战性,研究进展缓慢以及世界学术文化的变化,进入21世纪以来从事相关基础研究工作的化学研究者急剧减少.然而,毋庸置疑,实现温和条件下氮气的活化与转化是人类需要解决的重大科学问题,是人类社会可持续发展的要求,是科学家尤其是化学家最重要的使命之一.将氮气直接转化为含氮有机化合物是氮气的直接应用之一,本综述总结和归纳了文献报道的金属促进的以氮气为原料直接生成含氮有机化合物的转化方法和反应机理,产物主要包括胺类,酰胺类,酰亚胺类,腈类,二氮烯类,连氮类,碳二亚胺类,异氰酸酯类及杂环类有机化合物.本综述不包括将氮气转化成氨气和部分还原及质子化产物的文献.
English
Direct Transformation of N2 to N-Containing Organic Compounds
Abstract:
As a grand research area closely related to human civilization and living, the activation and transformation of dinitrogen (nitrogen fixation) under mild conditions used to be a central research theme worldwide in the 1970's~1990's. Nitrogen fixation is the process by which atmospheric nitrogen is directly converted to a bioavailable form. This basic chemical reaction process is essential to sustaining all life on this planet. However, due to great challenging of the nature of this research, slow progress and worldwide change of academic culture, the number of researchers engaged in this fundamental research area has been drastically reduced. Nevertheless, there is no doubt that realizing activation and transformation of dinitrogen under mild conditions is a grand scientific problem that people need to solve, required by sustainable development of human society. It is thus one of the most important missions of scientists, especially chemists. Three types of N-containing products can be obtained through direct transformation of dinitrogen. The most popular one is the formation of ammonia NH3 and NxHy. The industrial Haber-Bosch process, which requires harsh reaction conditions such as high temperature and pressure and uses at least 1%~2% of the annual primary energy supply in the world, is still the main method to produce ammonia from molecular dinitrogen and dihydrogen gases. Inspired by the investigation of nitrogenase and the discovery of the first molecular nitrogen complex in 1965, chemists have paid more attention to achieving the reduction of dinitrogen to ammonia with transition metal complexes either as regents or as catalysts. Reports on the other two types of products, the N-E (E=P, Si) bonding compounds, and the N-C bonding compounds, are very rare. Compared with ammonia, nitrogen-containing organic compounds such as amines, amides, imides, amino acids and aza-heterocycles are also high-value products. This review mainly summarizes the progress in the field of direct transformation of molecular nitrogen to nitrogen-containing organic compounds by using transition metal complexes, as well as the elucidation of transformation mechanisms. The N-containing organic compounds thus formed include amines, amides, imides, nitriles, diazenes, azines, carbodiimides, isocyanates and heterocycles. Although some progress has been achieved, examples are still very much limited, efficiency is generally very low. Transition metal complex-catalyzed reaction process is in great demand. Synergetic strategy is considered to be one of the efficient ways to realize transition metal complex-catalyzed direct transformation of molecular nitrogen to nitrogen-containing organic compounds under mild conditions. The formation of N-E (E=P, Si) bonding compounds and the reduction of dinitrogen to ammonia and other partially reduced or protonated products of dinitrogen are not covered here.
-
1 引言
氮是维持生命活动的必需元素, 是保障人类文明与生存的重要基础; 氮气是空气的最主要成分, 约占空气体积分数78%, 是最丰富、“最廉价”的氮源, 可谓取之不尽用之不竭.因此, 高效直接利用氮气, 使其真正成为最廉价的氮源, 一直是人类的梦想, 也一直是科学家尤其是化学家最重要的使命之一.
从氮气直接合成氨的工业合成氨技术(Haber-Bosch Process)是20世纪人类科学技术上的一项重大突破, 奠定了现代农业的基础, 极大地推动了现代文明的进步.但其苛刻的反应条件和超高能耗(据推算每年耗能量占全球耗能总量的1%~2%)使其难以符合人类社会可持续发展的要求.实现温和条件下氮气的直接高效活化与转化, 需要科学家探索和解决其中的基本科学问题, 是自然预设给科学家的“圣杯”.自1965年第一个过渡金属-氮气配合物被化学家合成出来之后[1], 模拟生物固氮模式的仿生化学固氮就成为国际上众多化学家包括我国化学家所采取的最主要研究方法[2, 3].随着近年来生物固氮酶FeMoco核心结构的日益明晰[4~10], 具有类似核心结构的过渡金属配合物被大量报道[3, 11].但是, 由于生物固氮酶具有复杂的组成与结构, 目前化学家仅仅可以模拟合成其组成的很小一部分, 因此这些人工合成的配合物还远远不能比拟其生物功能.虽然如此, 作为与人类文明和生存密切相关的重大研究方向, 化学家们从基础研究的角度已经取得了一些重要进展.
氮气的直接应用归纳起来可以生成三大类产物.如图 1所示, 一是生成氨以及氮气部分质子化产物NxHy, 二是生成含氮-硅或氮-膦键的产物, 三是生成含氮有机化合物(含有氮-碳键的化合物).目前已经取得的重要进展还主要集中在从氮气合成氨以及氮气部分质子化产物NxHy的研究领域[12~16].其中, 在均相化学研究领域, 实现温和条件下过渡金属催化的从氮气直接合成氨以及氮气部分质子化产物NxHy的报道屈指可数, 并且催化效率极低[17]; 另一个方面, 利用过渡金属配合物直接实现从氮气合成含氮-硅键[18~22]或氮-膦键[23]的产物以及含氮有机化合物的文献报道更少, 而生成氮-碳键的催化循环反应目前还没有实现[24].本文总结和归纳了文献报道的金属促进的以氮气为原料直接生成含氮有机化合物的转化方法和反应机理, 产物主要包括胺类, 酰胺类, 酰亚胺类, 腈类, 二氮烯类, 连氮类, 碳二亚胺类, 异氰酸酯类及杂环类有机化合物.本文不包括将氮气转化成含氮-硅或氮-膦键的产物以及氨气和部分还原及质子化产物的方法.
图 1
氮气转化领域的三个研究方向
Figure 1.
Research directions towards transformations of dinitrogen
2 以氮气为原料直接合成含氮有机化合物
2.1 合成胺类化合物
2.1.1 分子氮与金属有机化合物反应
1968年, Volpin课题组[25]报道了二茂二氯化钛Cp2TiCl2 (Cp=C5H5)或二茂二苯基钛Cp2TiPh2在PhLi存在的条件下均可促进氮气转化成苯胺和氨气的方法.研究发现当氮气的压力由常压增加到一百个大气压后, 苯胺和氨气的产率均明显升高.此外, 当使用钛配合物CpTiCl3, TiCl4或Ti (OBu)4时, 在PhLi参与下与氮气进行反应后经水解处理依然可以得到近似产率的苯胺.当用Cp2TiCl2与对甲基苯基锂在氮气压力为100个大气压的情况下进行反应后可以得到主产物对甲苯胺以及4%的间位异构体.在相同的反应条件下, 若用间甲基苯基锂进行反应可以得到产率为97%的间甲苯胺及产率接近3%的对位异构体, 其反应示意图如图 2所示.
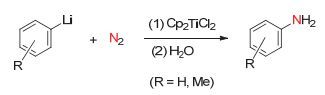 图 2
以苯基锂和氮气为原料制备苯胺
Figure 2.
Synthesis of aniline from phenyllithium and dinitrogen
图 2
以苯基锂和氮气为原料制备苯胺
Figure 2.
Synthesis of aniline from phenyllithium and dinitrogen
该研究组还提出了可能的反应机理(图 3), 金属钛前体与芳基锂反应首先形成钛配合物1, 接着氮气分子与之配位形成配合物2, 然后氮气分子进一步插入到金属钛-碳键形成二氮烯配合物3, 经过水解后得到芳香胺类产物.
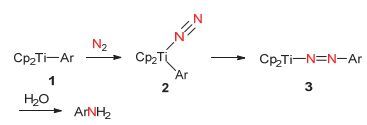 图 3
可能的反应机理
Figure 3.
Possible reaction mechanism
图 3
可能的反应机理
Figure 3.
Possible reaction mechanism
当氮气分子与邻甲苯基锂和Cp2TiCl2反应时, 意外地得到了三种甲基苯胺的异构体混合物:邻甲基苯胺(产率: 40%~77%), 间甲基苯胺(产率: 21%~52%), 对甲基苯胺(产率: 2%~8%).产生三种异构体的可能原因是邻甲基苯基锂与金属钛形成的苯炔中间体异构化所致.异构化的原因很可能因为邻位甲基的存在使得苯炔中间体位阻较大, 从而导致向位阻更小、结构更稳定的间位异构体转变.苯炔化合物的形成可以从Cp2TiCl2或Cp2TiPh2与PhLi反应产物中监测到三亚苯而得到证实.
该研究组还尝试将分子氮与Cp2TiCl2和脂肪族有机锂试剂(如n-BuLi)或格氏试剂(如C2H5MgBr)进行反应, 以期获得脂肪胺类产物.但脂肪胺类产物无法生成或产率极低.因此, 从PhLi到MeC6H4Li再到烷基锂, 其相应胺类产物的产率递减, 其相应的Ti—C键的稳定性也递减.该规律与胺类产物的生成机理相吻合, 即氮气分子插入到Ti—C键中是反应的关键步骤, Ti—C键在溶液中的存在时间越短, 氮气分子插入该键的可能性越低, 其相应的胺类产物的产率也越低.
2.1.2 分子氮与酮类化合物反应
Tamelen课题组[26]报道在Cp2TiCl2及镁粉存在的条件下, 酮及醛类化合物可以与氮气反应, 经过酸化处理后可以生成脂肪伯胺及脂肪仲胺类化合物.当提高胺化反应温度时, 产率略有降低.此外, 当使用萘钠作为还原剂时依然可以使该反应发生.酮类化合物包括二乙基酮, 二正丁基酮和环己酮, 醛类化合物包括苯甲醛, 其反应方程式见图 4.
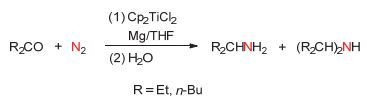 图 4
以酮及氮气为原料制备胺类化合物
Figure 4.
Synthesis of amines from ketones and dinitrogen
图 4
以酮及氮气为原料制备胺类化合物
Figure 4.
Synthesis of amines from ketones and dinitrogen
可能的反应机理如图 5所示, 首先羰基氧原子上的孤对电子进入钛金属的空轨道中, 实现氧与钛的配位.与此同时, 带负电荷的氮原子进攻羰基碳原子形成四元环状结构, 酸性条件下经四元开环得产物胺.该反应过程中的金属钛是至关重要的, 若形成其他金属氮化物将不能实现碳原子的氨基化反应.
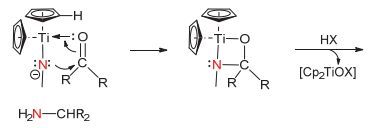 图 5
可能的反应机理
Figure 5.
Possible reaction mechanism
图 5
可能的反应机理
Figure 5.
Possible reaction mechanism
2.1.3 分子氮与芳香卤代烃反应
Mori课题组[27]于1998年报道了以芳香卤代烃为原料, 在钛氮配合物和零价钯参与的情况下通过转金属化作用而合成芳香伯胺及芳香仲胺的方法.该方法首先利用Ti (OiPr)4, Li及TMSCl与氮气分子形成的钛氮配合物(Ti-N Complexes)作为反应原料, 并将钛氮配合物中的Ti-N (TMS)3作为关键反应活性中间体, 通过转金属化作用将钛氮配合物中与钛原子相连的氮源部分转移到零价钯原子上, 从而完成最终的转化.以对苯基溴苯为例, 在零价钯配合物Pd2(dba)3和膦配体P (o-tolyl)3存在的条件下, 将其与钛氮配合物进行反应, 可以成功得到芳香伯胺化合物及芳香仲胺化合物.该反应的溶剂为甲苯, 若换成四氢呋喃则无法得到相应产物.此外, 延长该反应的时间有利于仲胺产物的生成.不同膦配体对反应产物的影响较大, 实验结果表明用(S)-BINAP双膦配体参与反应, 能以较高的产率得到芳香伯胺主产物, 若改用其他双膦配体如DPPF, DPPP及(S)-BINAPO参与反应, 则无法形成仲胺产物.因此, 双膦配体参与该反应将以芳香伯胺产物为主.
苯环上带有不同取代基的溴苯也适应此类反应, 当苯环上有给电子基团时产物以仲胺为主, 当苯环上有吸电子基团时产物以伯胺为主.值得注意的是, 当溴原子的邻位引入两个甲基时, 该反应并没有因空间阻碍而受到影响, 仍能以47%的产率得到芳香伯胺产物, 以16%的收率得到芳香仲胺产物(图 6).
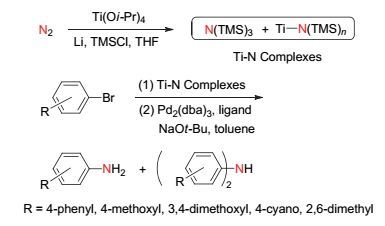 图 6
以芳香卤代烃和氮气为原料制备芳香胺类产物
Figure 6.
Synthesis of aromatic amines from aromatic halohydrocarbon and dinitrogen
图 6
以芳香卤代烃和氮气为原料制备芳香胺类产物
Figure 6.
Synthesis of aromatic amines from aromatic halohydrocarbon and dinitrogen
作者提出了此类反应的关键活性中间体.当用N (TMS)3或HN (TMS)2代替钛氮配合物时, 无法得到任何含氮产物, 这充分证明了此类反应的活性中间体是钛氮配合物中的Ti-N (TMS)n, 而不是N (TMS)3.此外, 与钛原子相连的氮源部分从金属钛转移到金属钯是反应的关键步骤, 其可能的反应历程如图 7所示.
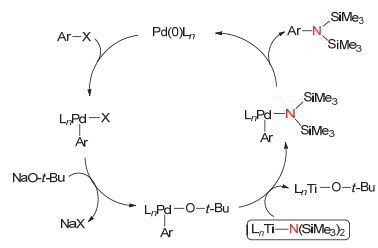 图 7
可能的反应历程
Figure 7.
Possible reaction mechanism
图 7
可能的反应历程
Figure 7.
Possible reaction mechanism
2.2 合成酰胺及酰亚胺类化合物
2.2.1 分子氮与酰卤反应
Mori课题组[28]以芳香酰氯为原料, 在TiCl3及金属Mg存在的条件下, 于氮气环境下合成出芳香酰胺与芳香酰亚胺.反应过程如下, 首先TiCl3在四氢呋喃中与金属Mg和氮气分子形成配合物[TiNMg2Cl2•THF], 然后该配合物进一步与芳香酰氯反应, 经过水解后得到相应的芳香酰胺及芳香酰亚胺产物(图 8).
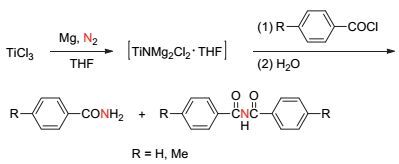 图 8
以芳香酰氯和氮气为原料制备芳香酰胺及酰亚胺
Figure 8.
Synthesis of aromatic amides and imides from aroyl chloride and dinitrogen
图 8
以芳香酰氯和氮气为原料制备芳香酰胺及酰亚胺
Figure 8.
Synthesis of aromatic amides and imides from aroyl chloride and dinitrogen
若用TiCl4, Cp2TiCl2或VCl3替代TiCl3, 也能得到相应的酰胺及酰亚胺产物.升高反应温度及增加芳香酰氯的用量均有利于芳香酰胺及芳香酰亚胺产率的提高.该课题组还尝试将钛氮配合物[TiNMg2Cl2•THF]与不饱和酰氯进行反应, 发现可以得到相应的酰胺产物(图 9).但是当其与相应结构的饱和酰氯进行反应却不能得到酰胺产物.这主要是因为不饱和双键的存在可以与酰氯中的羰基形成共轭效应从而起到稳定羰基的作用, 而饱和酰氯则没有该作用, 其羰基容易发生脱羰反应, 因此无法形成酰胺产物.
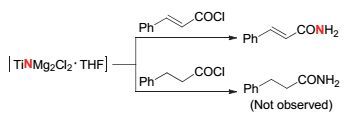 图 9
以不饱和酰氯来制备酰胺
Figure 9.
Synthesis of amides from unsaturated acyl chloride
图 9
以不饱和酰氯来制备酰胺
Figure 9.
Synthesis of amides from unsaturated acyl chloride
Mori等还推测了其生成酰胺及酰亚胺的机理(图 10), 若酰氯与钛氮配合物按照酰胺配合物的方式结合, 其水解后将得到酰胺产物.若酰氯与钛氮配合物按照酰亚胺配合物的方式结合, 其水解后将得到酰亚胺产物.
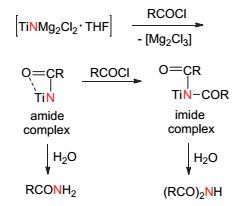 图 10
可能的反应机理
Figure 10.
Possible reaction mechanism
图 10
可能的反应机理
Figure 10.
Possible reaction mechanism
2.2.2 分子氮与芳香卤代烃和一氧化碳反应
Mori课题组[29]于2000年报道了以钛氮配合物(Ti-N Complexes)和芳基卤代烃为原料, 在一氧化碳氛围中通过转金属化作用而制备芳香酰胺的方法.在Pd2(dba)3及膦配体DPPF和叔丁醇钠存在下, 将对位含有甲氧甲基的溴苯与钛氮配合物和一氧化碳反应后, 经水解处理分别得到产率为4%的芳香酰胺, 1%的芳香酰亚胺, 6%的芳香腈以及16%的芳香伯胺产物.虽然前三种产物的产率较低, 但是该反应已成功将CO及N2在温和条件下引入到芳香卤代烃中(图 11).当用DMF代替四氢呋喃作为溶剂时, 产物芳香酰胺及酰亚胺的产率可分别提高到24%与11%, 若用丙腈作为溶剂时, 芳香酰胺的产率可以提高到47%, 若用BINAP配体代替DPPF, 用甲苯作为溶剂时, 产物以芳香腈为主, 产率可达56%.
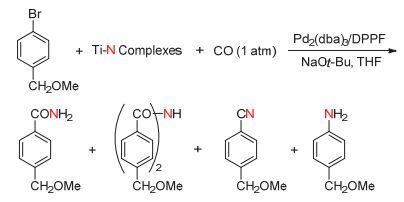 图 11
以芳香卤代烃和一氧化碳为原料制备酰胺及酰亚胺
Figure 11.
Synthesis of aromatic amides and imides from aromatic halohydrocarbon and carbon monoxide
图 11
以芳香卤代烃和一氧化碳为原料制备酰胺及酰亚胺
Figure 11.
Synthesis of aromatic amides and imides from aromatic halohydrocarbon and carbon monoxide
其可能的反应机理如图 12所示, 首先芳香卤代烃和零价钯发生氧化加成反应生成ArPdLnX, 并在CO参与下形成酰基钯配合物ArCOPdLnX, 该配合物可与钛氮配合物(Ti-N Complexes)发生转金属化反应, 即钛氮配合物中的氮原子转移到金属钯原子上, 从而生成中间体A或中间体B, 中间体A可形成由Ti及Pd组成的五元环状结构, 该五元环可发生还原消除反应, 从而释放出零价钯并形成包含金属Ti在内的四元环结构, 该四元环可进一步转化成腈类产物.此外, 中间体A也可与溶剂DMF形成四元环结构, 该四元环结构可进一步转变成亚胺, 最后经水解得酰胺产物, 而中间体B则可直接经水解反应生成酰胺产物.通常情况下, 由芳香卤代烃合成芳香酰胺, 要由伯胺或仲胺在金属钯存在及一氧化碳环境下进行亲核取代反应而完成, 此时产物中的氮原子会有取代基.若要氮原子上不含取代基, 则需要用氨气作为亲核试剂, 但氨气的亲核性较低, 无法完成此反应.因此, Mori组报道的此类反应的一个显著特点在于, 用该合成方法可以有效地合成氮原子无取代基的芳香酰胺类化合物.
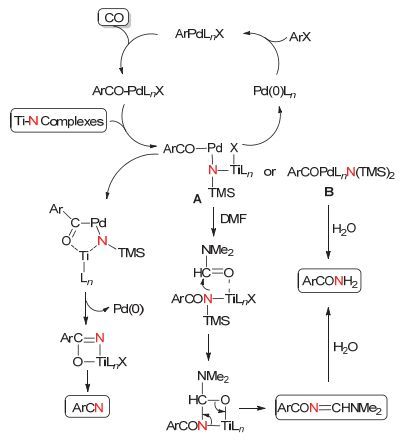 图 12
可能的反应历程
Figure 12.
Possible reaction mechanism
图 12
可能的反应历程
Figure 12.
Possible reaction mechanism
2.2.3 分子氮与过渡金属配合物和一氧化碳反应
Chirik课题组[30]于2010年报道了以金属铪的氮气配合物为原料合成草酰胺的方法(图 13).该方法首先以铪的二氯化物Hf-Cl2为起始原料, 将其与过量的三甲基碘化硅反应得到相应的二碘化物Hf-I2, 该配合物在钠汞齐作还原剂的条件下, 可与氮气分子配位形成金属铪氮气配合物Hf-N2, 此氮气配合物可以进一步与CO发生插入反应, 两分子CO可分别与两个氮原子通过碳氮双键相连, 从而形成配合物Hf-N2C2O2, 并在过量盐酸的作用下将草酰胺从配合物骨架中释放出来.该方法可实现在温和条件下对惰性小分子氮气与CO同时活化的目的, 并最终将其转化到有机化合物中.
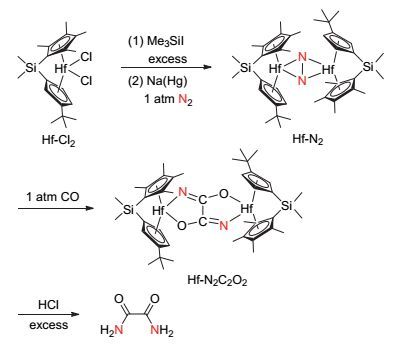 图 13
以氮气为原料合成草酰胺
Figure 13.
Synthesis of oxamide from dinitrogen
图 13
以氮气为原料合成草酰胺
Figure 13.
Synthesis of oxamide from dinitrogen
2.3 合成腈类化合物
2.3.1 分子氮与酰卤反应
Cummins课题组[31]于2006年报道了以氮气和金属钼配合物为原料来合成腈类化合物的方法, 该方法可以构建合成循环, 即金属钼配合物经过还原处理后可以重新生成, 整个合成反应循环过程如图 14所示.氮气分子在NaH存在下与金属钼配合物Mo[N (t-Bu) Ar]3反应生成含有钼氮三键的配合物N≡Mo[N (t-Bu) Ar]3, 该配合物进一步与酰氯化合物反应可以构建新的碳氮键, 然后经过金属镁及Me3SiOTf处理后形成碳氮双键, 再经过氯化锡或氯化锌的处理即可将腈类产物从金属配合物中释放出来.在生成腈类产物的同时, 还会产生氯原子与金属钼配位的配合物Cl-Mo[N (t-Bu) Ar]3, 该配合物再与金属镁反应后可以重新生成反应原料Mo[N (t-Bu) Ar]3, 从而完成整个合成循环过程.钼氮三键配合物N≡Mo[N (t-Bu) Ar]3是一个弱的亲核试剂, 该配合物不能直接与酰氯化合物进行反应, 必须在适当添加剂存在下才能完成反应, 这可能是该配合物中氮原子周围存在三个大位阻的叔丁基所致.值得注意的是, 如果将ZnCl2换成ZnI2或Zn (OTf)2, 反应将不能生成腈类产物, 这表明Mo—Cl键的形成是释放出腈类产物的关键步骤.
 图 14
以氮气为原料合成有机腈类化合物的反应循环图
Figure 14.
A synthetic cycle that incorporates dinitrogen into organic nitriles
图 14
以氮气为原料合成有机腈类化合物的反应循环图
Figure 14.
A synthetic cycle that incorporates dinitrogen into organic nitriles
侯召民课题组[32]于2016年报道了以氮气分子与金属钛形成的四钛金属配合物为原料合成腈类化合物的方法(图 15).四钛金属配合物可与各种芳香酰氯或脂肪酰氯在溶剂苯中进行反应, 从而生成相应的芳香腈或脂肪腈类产物.该方法的反应底物适应性较强, 苯环上连有卤素取代基, 硝基取代基, 氯甲基以及醛基等官能团时, 反应均不受影响.此外, 该方法反应条件温和, 不需要添加其他反应试剂.
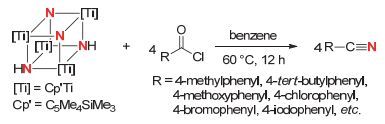 图 15
以氮气和酰氯为原料合成腈类化合物
Figure 15.
Synthesis of nitrile from dinitrogen and acyl chloride
图 15
以氮气和酰氯为原料合成腈类化合物
Figure 15.
Synthesis of nitrile from dinitrogen and acyl chloride
2.3.2 分子氮与过渡金属配合物和一氧化碳反应
Chirik课题组[33]报道了以金属铪氮气配合物为原料来合成乙腈的方法(图 16).金属铪氮气配合物Hf-N2可与CO分子发生插入反应形成四金属中心的配合物Hf-N4C4O4, 该配合物在110 ℃条件下发生解离从而形成双金属中心配合物, 并同时生成与金属铪配位的氰基配体, 氰基配体可与三氟甲磺酸甲酯作用并同时从配合物骨架中分离, 从而生成乙腈.虽然该反应并没有催化循环活性, 但产物中氰基官能团的氮原子与碳原子分别来自于氮气分子与一氧化碳分子, 由此可见该反应能同时活化两种惰性气体小分子并可将其成功转化到有机化合物中.
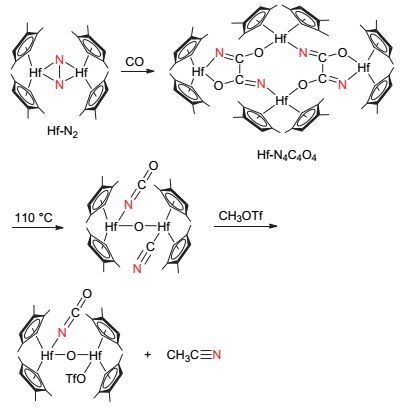 图 16
以氮气和一氧化碳为原料合成乙腈
Figure 16.
Synthesis of acetonitrile from dinitrogen and carbon monoxide
图 16
以氮气和一氧化碳为原料合成乙腈
Figure 16.
Synthesis of acetonitrile from dinitrogen and carbon monoxide
2.3.3 分子氮与过渡金属配合物反应
Schneider课题组[34]于2016年报道了以氮气和含有PNP配体的金属铼氯化物为原料合成乙腈的方法(图 17).首先金属铼氯化物在钠汞齐作还原剂的条件下可与氮气反应生成含有铼氮三键的配合物, 该配合物与三氟甲磺酸乙酯作用后可形成含有铼氮双键和碳氮双键的配合物, 其进一步与N-氯代琥珀酰亚胺反应后, 能以93%的产率将乙腈从配合物体系中释放出来, 并同时重新生成原料金属铼氯化物, 从而完成整个合成循环过程.
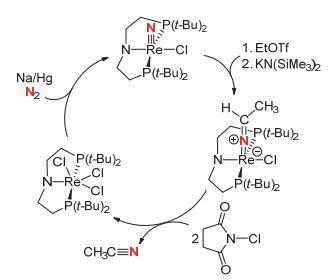 图 17
以氮气为原料合成乙腈的反应循环图
Figure 17.
A synthetic cycle that incorporates dinitrogen into acetonitrile
图 17
以氮气为原料合成乙腈的反应循环图
Figure 17.
A synthetic cycle that incorporates dinitrogen into acetonitrile
2.4 合成二氮烯类化合物
2.4.1 分子氮与金属有机化合物反应
Weiss课题组[35]报道了首例具有潜在催化循环特征的金属配合物催化氮气向含氮有机化合物转化的反应体系(图 18).以氮气与金属锰所形成的配合物4为反应原料, 将其与甲基锂反应得到氮甲基化的配合物5, 5与可提供甲基正离子的(CH3)3OBF4反应, 得到双氮甲基化配合物6, 6在100 bar的氮气压力下可以释放出二甲基二氮烯, 并重新生成配合物4, 从而完成催化循环过程.该循环过程中的副反应会使催化剂很快失去催化活性, 因此该循环过程并没有太大的实际应用价值, 但此过程是首例具有催化循环潜能的反应, 可为今后研究由氮气直接合成含氮有机化合物提供较大的参考.
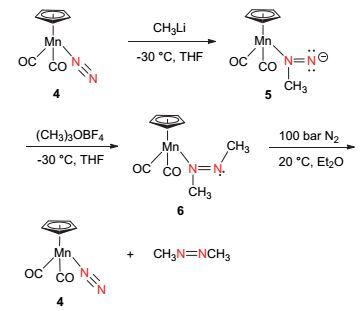 图 18
以氮气为原料合成二氮烯类化合物
Figure 18.
A synthetic cycle that incorporates dinitrogen into diazenes
图 18
以氮气为原料合成二氮烯类化合物
Figure 18.
A synthetic cycle that incorporates dinitrogen into diazenes
2.5 合成连氮类化合物
2.5.1 分子氮与过渡金属配合物和酮类化合物反应
Hidai课题组[36]报道了以氮气和金属钨配合物cis-[W (N2)2(PMe2Ph)4]为原料合成连氮类化合物的方法.反应体系在甲醇, 乙醇或水存在下, 配合物cis-[W (N2)2-(PMe2Ph)4]可与酮类化合物反应而生成相应的连氮类产物.酮类化合物包括丙酮, 甲基乙基酮, 二乙基酮, 苯基甲基甲酮和二苯甲酮.当用二苯甲酮进行该反应时, 反应体系中有中等产率的二苯甲酮腙和二苯甲酮连氮两种产物.这表明此类反应的终产物连氮类化合物的形成先经历了腙类化合物, 由腙类产物进一步转化而来, 其反应过程如图 19所示.
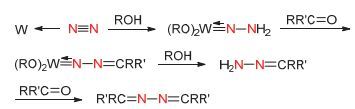 图 19
以氮气为原料合成连氮类化合物
Figure 19.
Synthesis of azine from dinitrogen
图 19
以氮气为原料合成连氮类化合物
Figure 19.
Synthesis of azine from dinitrogen
2.5.2 分子氮与过渡金属配合物和二氧化碳反应
Chirik课题组[37]利用氮气与铪的配合物Hf-N2合成出氮原子上有三甲基硅基和酯基取代的连氮类化合物(图 20).金属铪的分子氮配合物Hf-N2与两分子的二氧化碳进行反应, 可成功将二氧化碳分子插入到Hf与N之间, 从而成功构建新的碳氮键.在过量Me3SiI存在的条件下, 碳氮键将与金属配合物骨架分离, 从而得到连氮类产物和铪的二碘化物Hf-I2.虽然最终的连氮产物尚无法从体系中分离, 但核磁与气质联用技术已确证该产物的形成.值得注意的是, 在生成连氮产物的同时, 体系可将金属铪配合物释放出来, 若能找到其与氮气再次配位的条件, 该反应有可能形成催化循环.
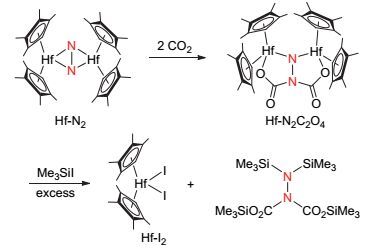 图 20
以氮气和二氧化碳为原料合成连氮类化合物
Figure 20.
Synthesis of azine from dinitrogen and carbon dioxide
图 20
以氮气和二氧化碳为原料合成连氮类化合物
Figure 20.
Synthesis of azine from dinitrogen and carbon dioxide
Chirik课题组[38]又在此研究结果的基础上, 将金属铪氮气配合物换成金属锆氮气配合物, 同样可以得到类似的连氮类产物(图 21).首先金属锆氮气配合物Zr-N2与二氧化碳发生插入反应形成碳氮键, 与上述反应不同之处在于, 该插入反应并未使两分子二氧化碳与同一个氮原子相连, 而是与两个氮原子分别相连, 形成产物Zr-N2C2O4, 该配合物与过量的三甲基碘化硅反应可以得到氮原子有三甲基硅基取代的连氮产物, 若与三氟甲磺酸甲酯反应后经水解处理可得双甲氧羰基取代的连氮产物.此类金属氮气配合物不仅可以活化惰性的氮气分子, 还可同时活化二氧化碳分子, 并使其成功转化到有机化合物中.
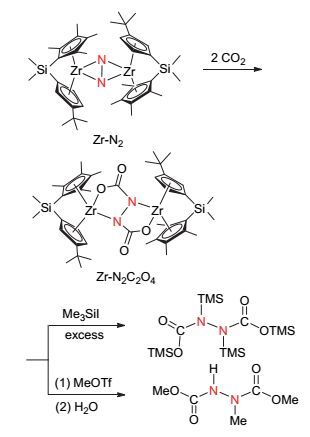 图 21
以氮气和二氧化碳为原料合成连氮类化合物
Figure 21.
Synthesis of azine from dinitrogen and carbon dioxide
图 21
以氮气和二氧化碳为原料合成连氮类化合物
Figure 21.
Synthesis of azine from dinitrogen and carbon dioxide
2.6 合成碳二亚胺类化合物
2.6.1 分子氮与过渡金属配合物和一氧化碳反应
Chirik课题组[39]于2013年首次报道了以金属铪氮气配合物为原料来合成碳二亚胺的方法(图 22).金属铪氮气配合物Hf-N2可与CO分子发生插入反应形成含有碳氮双键及铪氮双键的配合物Hf-N2CO, 该配合物可进一步与叔丁基异氰酸酯作用, 使异氰酸酯中的碳与氮形成第二个碳氮双键, 从而得到配合物Hf-N3C2O2, 该配合物在120 ℃条件下会发生分子内重排反应, 使异氰酸酯中的碳氧键断裂, 并同时形成碳二亚胺结构配体, 该配体在三氟甲磺酸甲酯的作用下可从金属配合物骨架中解离, 从而生成碳二亚胺类产物, 该产物中的一个氮原子来自于氮气分子, 另一个氮原子则来自于叔丁基异氰酸酯.
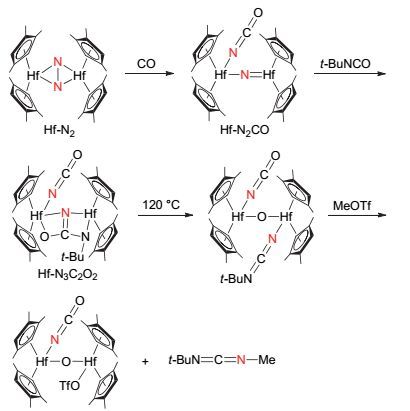 图 22
以氮气和一氧化碳为原料合成碳二亚胺类化合物
Figure 22.
Synthesis of carbodiimide from dinitrogen and carbon monoxide
图 22
以氮气和一氧化碳为原料合成碳二亚胺类化合物
Figure 22.
Synthesis of carbodiimide from dinitrogen and carbon monoxide
2.7 合成异氰酸酯类化合物
2.7.1 分子氮与过渡金属配合物和二氧化碳反应
Sita课题组[40]报道了以金属钼或钨的氮气配合物为反应原料来合成异氰酸酯类化合物的方法(图 23).首先钼或钨的氯化物在金属钠作为还原剂的条件下, 可与氮气配位形成双金属氮气配合物, 该配合物在光照条件下与三甲基氯硅烷进行反应后, 可将硅原子引入到氮原子上并同时形成金属氮双键, 该配合物进一步与二氧化碳作用后可释放出异氰酸酯类产物Me3SiNCO, 同时氧原子与金属相连形成含有金属氧双键的配合物, 该配合物与三甲基氯硅烷反应后可以重新生成起始原料金属氯化物, 从而完成整个合成循环过程, 但此反应尚不具备催化循环能力.
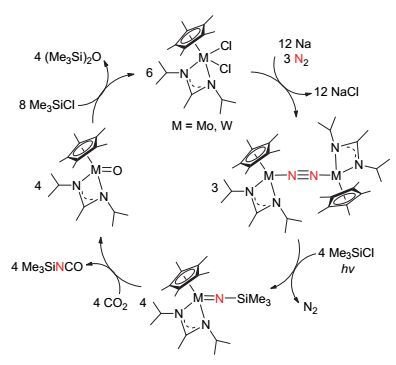 图 23
以氮气为原料合成异氰酸酯类化合物的反应循环图
Figure 23.
A synthetic cycle that incorporates dinitrogen into isocyanate
图 23
以氮气为原料合成异氰酸酯类化合物的反应循环图
Figure 23.
A synthetic cycle that incorporates dinitrogen into isocyanate
2.8 合成含氮杂环类化合物
2.8.1 分子氮与过渡金属配合物和羰基化合物反应
Hidai课题组[41]报道了以氮气和金属钼或金属钨配合物为原料来合成吡咯和N-氨基吡咯的方法(图 24).该方法以氮气分子与金属钼或钨结合形成的配合物1a或1b为起始原料, 将其与盐酸或四氟硼酸反应可以得到含有金属氮三键的配合物2a~2c, 该配合物进一步与2, 5-二甲氧基四氢呋喃反应可以将吡咯环引入到配合物体系内, 然后经过四氢铝锂的还原后可以将吡咯环与金属配合物体系分离, 从而最终合成出吡咯化合物及N-氨基吡咯化合物.该反应还会生成氨气和金属四氢化物, 该金属四氢化物可以在氮气及钨灯光照条件下释放出氢气并重新结合氮气而生成原料配合物1a或1b, 从而完成催化循环过程(图 25).
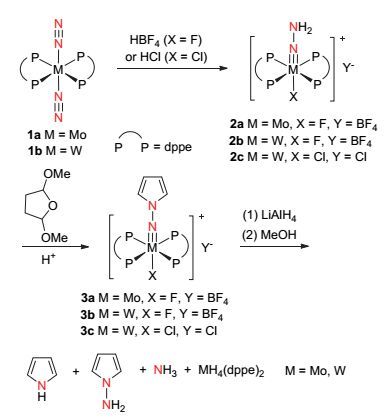 图 24
以氮气为原料合成吡咯和N-氨基吡咯
Figure 24.
Synthesis of pyrrole and N-amino pyrrole from dinitrogen
图 24
以氮气为原料合成吡咯和N-氨基吡咯
Figure 24.
Synthesis of pyrrole and N-amino pyrrole from dinitrogen
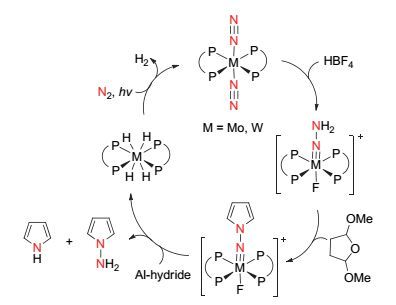 图 25
以氮气为原料合成吡咯和N-氨基吡咯的催化循环过程
Figure 25.
A synthetic cycle that incorporates N2 into pyrrole and N-amino pyrrole
图 25
以氮气为原料合成吡咯和N-氨基吡咯的催化循环过程
Figure 25.
A synthetic cycle that incorporates N2 into pyrrole and N-amino pyrrole
利用该方法, 可以合成β位含有取代基的吡咯化合物(图 26).将含有吡咯环结构的配合物3c作为起始原料, 在三氯化铝存在的条件下, 与正庚酰氯发生傅克酰基化反应, 将正庚酰基引入到吡咯环的β位上, 再用四氢铝锂还原金属配合物, 使β位有取代基的吡咯化合物从配合物中解离, 实现在吡咯环β位上引入官能团的目的, 而此类吡咯环衍生物用一般的化学方法是难以合成的.
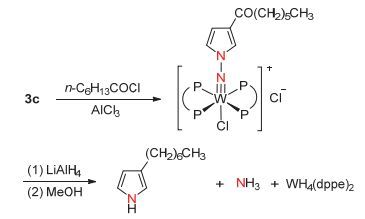 图 26
以氮气为原料合成β位有取代基的吡咯
Figure 26.
Synthesis of pyrrole with a substituent on the β position from dinitrogen
图 26
以氮气为原料合成β位有取代基的吡咯
Figure 26.
Synthesis of pyrrole with a substituent on the β position from dinitrogen
Mori课题组[42]于1995年报道了以氮气和二羰基或三羰基化合物为原料合成含氮杂环有机化合物的方法, 该方法以钛氮配合物Ti-N Complexes [N (TMS)3 and Ti-N (TMS)n]作为活性中间体, 将其与相应的羰基化合物进行反应, 从而实现目标产物的转化.该方法与Paal-Knorr合成吡咯法[43]比较类似, 不同之处在于Paal-Knorr是用氨气或伯胺作为氮源, 而该方法是用钛氮配合物作为氮源.值得注意的是, 当反应底物环酮的β位含有OH或OTf时, 产物可以保留其中一个羰基, 这是与Paal-Knorr合成法显著不同之处(图 27), 当羰基处于1位和4位且β位不含OTf或OH时, 则生成与Paal-Knorr合成法一致的产物(图 28).该合成方法的另一个显著特点是可以将三羰基化合物转化成含氮杂环产物, 这是Paal-Knorr合成吡咯法不能实现的反应(图 29).
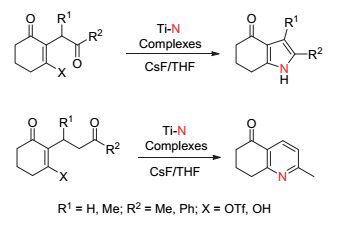 图 27
以氮气为原料合成吲哚及喹啉类衍生物
Figure 27.
Synthesis of derivatives of indole and quinoline from dinitrogen
图 27
以氮气为原料合成吲哚及喹啉类衍生物
Figure 27.
Synthesis of derivatives of indole and quinoline from dinitrogen
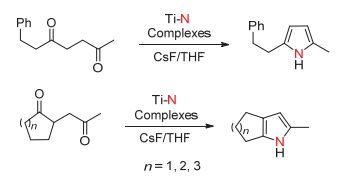 图 28
以氮气为原料合成吡咯类衍生物
Figure 28.
Synthesis of derivatives of pyrrole from dinitrogen
图 28
以氮气为原料合成吡咯类衍生物
Figure 28.
Synthesis of derivatives of pyrrole from dinitrogen
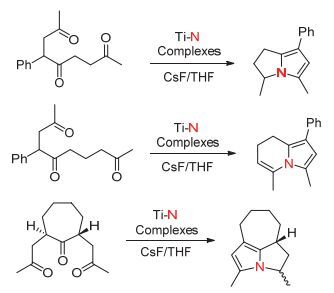 图 29
以氮气为原料合成吡呤类及吲嗪类衍生物
Figure 29.
Synthesis of derivatives of pyrrolizine and indolizine from dinitrogen
图 29
以氮气为原料合成吡呤类及吲嗪类衍生物
Figure 29.
Synthesis of derivatives of pyrrolizine and indolizine from dinitrogen
该反应体系能以氮气为原料, 并通过一锅法的方式将羰基化合物成功转化成一系列含氮杂环有机化合物, 其中包括吲哚衍生物, 喹啉衍生物, 吡咯衍生物, 吡呤衍生物以及吲嗪衍生物.值得注意的是, 在该反应体系中如果金属锂和三甲基氯硅烷过量存在, 则TiCl4可以作为催化剂而建立催化循环系统.
Mori课题组[44]还将反应底物扩展到酯类化合物, 当形成吡咯环时酯基未受到影响, 可见此类反应有较好的官能团耐受性.若用磷酸酯作为反应底物时, 产物将转化成环酰胺类化合物(图 30).此外, 当用空气代替氮气来进行此类反应时, 依然可以得到目标产物.虽然用氮气作为氮源时, 目标产物的产率基本上都高于以空气作为氮源时的产率, 但是以空气直接作为氮源的方法有更实际的应用前景.
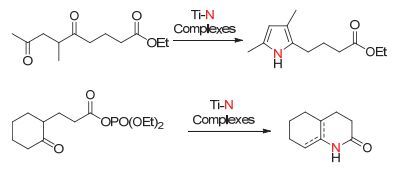 图 30
以氮气和酯类化合物为原料合成吡咯类衍生物及环酰胺类化合物
Figure 30.
Synthesis of derivatives of pyrrole and cyclic amide compounds from dinitrogen and ester compounds
图 30
以氮气和酯类化合物为原料合成吡咯类衍生物及环酰胺类化合物
Figure 30.
Synthesis of derivatives of pyrrole and cyclic amide compounds from dinitrogen and ester compounds
除了本文上述的用化学合成方法将氮气直接引入到含氮有机化合物中外, 还可利用电化学方法将氮气转化成含氮有机化合物.例如, 1981年Pickett等[45]就曾报道以金属钼的氮气配合物trans-[Mo (N2)2(dppe)2]和溴代烃为原料来合成烷基取代肼的方法.虽然用电化学方法固氮的报道目前还极其稀少, 但该方法或许可为今后固氮领域的研究开辟一条新的道路.
3 总结与展望
氮气的活化与转化研究是人们长久以来关注的研究热点, 科学家们一直期待能用化学方法模拟生物固氮过程, 并能在常温常压或相对温和的条件下, 将空气中的氮固定并使其转化成具有更高利用价值的含氮有机化合物.因此, 研究温和条件下氮气的固定和高效转化无疑是合成科学乃至人类面临的最具挑战性的重大基础研究课题.但令人遗憾的是, 近几十年来以氮气为原料直接合成含氮有机化合物的文献报道并不多见, 而具有催化循环活性的反应体系更是微乎其微.因此, 氮气活化与转化研究领域的核心科学问题依然是如何在具有高效催化循环能力的反应体系中构建新的C—N键.
将过渡金属配合物作为活化及转化氮气的催化剂很可能是未来长期研究的重点方向, 目前文献采取的方法是先将氮气与金属形成配合物, 再将配合物与有机部分结合, 最后经过水解等处理步骤而得到终产物.未来氮气活化与转化研究领域的发展趋势将主要集中在配体的合理设计方面, 例如将结构新颖的配体与适当的过渡金属进行合理的搭配, 高效的“协同效应”很可能是未来发现高效催化剂体系的关键突破口, 这是因为配体结构的微小变化, 往往容易导致配位金属中心周围电子云密度及空间位阻的变化, 这些变化可以进一步影响其活化氮气的效果, 从而有望实现高效转化氮气的终极目标.此外, 随着固氮酶结构的日益明晰, 以其催化活性中心为指导原则, 重点研究铁或钼等单核金属配合物对氮气的活化, 以及考察多核金属配合物通过协同作用对氮气进行活化的机理将会使氮气活化与转化研究领域达到更深入和更崭新的阶段.
-
-
[1]
Allen, A. D.; Senoff, C. V. Chem. Commun. 1965, 621.
-
[2]
(a) Jin, D. Chem. Bull. 1979, 42, 17(in Chinese). (金斗满, 化学通报, 1979, 42, 17. ) (b) Xi, Z. ; Jin, D. Chem. Bull. 1991, 54, 1(in Chinese). (席振峰, 金斗满, 化学通报, 1991, 54, 1. ) (c) Ma, X. ; Lei, M. Prog. Chem. 2013, 25, 1325(in Chinese). (马雪璐, 雷鸣, 化学进展, 2013, 25, 1325. ) (d) Zhang, Y. ; Ma, X. ; Zhang, X. ; Lei, M. Acta Chim. Sinica 2016, 74, 340(in Chinese). (张益伟, 马雪璐, 张欣, 雷鸣, 化学学报, 2016, 74, 340. ) (e) Yue, G. ; Gao, R. ; Zhao, P. ; Chu, M. ; Shuai, M. Acta Chim. Sinica 2016, 74, 657(in Chinese). (岳国宗, 高瑞, 赵鹏翔, 褚明福, 帅茂兵, 化学学报, 2016, 74, 657. )
-
[3]
(a) Chatt, J.; Dilworth, J. R.; Richards, R. L. Chem. Rev. 1978, 78, 589. (b) Hidai, M.; Mizobe, Y. Chem. Rev. 1995, 95, 1115. (c) Hidai, M. Coord. Chem. Rev. 1999, 185, 99. (d) Fryzuk, M. D.; Johnson, S. A. Coord. Chem. Rev. 2000, 200, 379. (e) Shaver, M. P.; Fryzuk, M. D. Adv. Synth. Catal. 2003, 345, 1061. (f) Fryzuk, M. D. Chem. Rec. 2003, 3, 2. (g) MacKay, B. A.; Fryzuk, M. D. Chem. Rev. 2004, 104, 385. (h) Crossland, J. L.; Tyler, D. R. Coord. Chem. Rev. 2010, 254, 1883. (i) Hazari, N. Chem. Soc. Rev. 2010, 39, 4044. (j) Tanabe, Y.; Nishibayashi, Y. Coord. Chem. Rev. 2013, 257, 2551. (k) van der Ham, C. J. M.; Koper, M. T. M.; Hetterscheid, D. G. H. Chem. Soc. Rev. 2014, 43, 5183. (l) Lehnert, N.; Peters, J. C. Inorg. Chem. 2015, 54, 9229. (m) Walter, M. D. Adv. Organomet. Chem. 2016, 65, 261. (n) Bhattacharya, P.; Prokopchuk, D. E.; Mock, M. T. Coord. Chem. Rev. 2017, 334, 67. (o) Burford, R. J.; Yeo, A.; Fryzuk, M. D. Coord. Chem. Rev. 2017, 334, 84.
-
[4]
Howard, J. B.; Rees, D. C. Chem. Rev. 1996, 96, 2965. doi: 10.1021/cr9500545
-
[5]
Einsle, O.; Tezcan, F. A.; Andrade, S. L. A.; Schmid, B.; Yoshida, M.; Howard, J. B.; Rees, D. C. Science 2002, 297, 1696. doi: 10.1126/science.1073877
-
[6]
Spatzal, T.; Aksoyoglu, M.; Zhang, L.; Andrade, S. L. A.; Schleicher, E.; Weber, S.; Rees, D. C.; Einsle, O. Science 2011, 334, 940. doi: 10.1126/science.1214025
-
[7]
Lancaster, K. M.; Roemelt, M.; Ettenhuber, P.; Hu, Y.; Ribbe, M. W.; Neese, F.; Bergmann, U.; DeBeer, S. Science 2011, 334, 974. doi: 10.1126/science.1206445
-
[8]
Lancaster, K. M.; Hu, Y.; Bergmann, U.; Ribbe, M. W.; DeBeer, S. J. Am. Chem. Soc. 2013, 135, 610. doi: 10.1021/ja309254g
-
[9]
Wiig, J. A.; Hu, Y.; Lee, C. C.; Ribbe, M. W. Science 2012, 337, 1672.
-
[10]
(a) Chen, Y.; Liu, L.; Peng, Y.; Chen, P.; Luo, Y.; Qu, J. J. Am. Chem. Soc. 2011, 133, 1147. (b) Luo, Y.; Li, Y.; Yu, H.; Zhao, J.; Chen, Y.; Hou, Z.; Qu, J. Organometallics 2012, 31, 335. (c) Li, Y.; Li, Y.; Wang, B.; Luo, Y.; Yang, D.; Tong, P.; Zhao, J.; Luo, L.; Zhou, Y.; Chen, S.; Cheng, F.; Qu, J. Nat. Chem. 2013, 5, 320. (d) Ouyang, Z.; Cheng, J.; Li, L.; Bao, X.; Deng, L. Chem. Eur. J. 2016, 22, 14162.
-
[11]
(a) Shima, T.; Hu, S.; Luo, G.; Kang, X.; Luo, Y.; Hou, Z. Science 2013, 340, 1549. (b) MacLeod, K. C.; Vinyard, D. J.; Holland, P. L. J. Am. Chem. Soc. 2014, 136, 10226. (c) Rittle, J.; McCrory, C. C. L.; Peters, J. C. J. Am. Chem. Soc. 2014, 136, 13853. (d) Ishida, Y.; Kawaguchi, H. J. Am. Chem. Soc. 2014, 136, 16990. (e) Lee, Y.; Sloane, F. T.; Blondin, G.; Abboud, K. A.; García-Serres, R.; Murray, L. J. Angew. Chem. Int. Ed. 2015, 54, 1499. (f) Pappas, I.; Chirik, P. J. J. Am. Chem. Soc. 2015, 137, 3498.
-
[12]
Nishibayashi, Y. Inorg. Chem. 2015, 54, 9234. doi: 10.1021/acs.inorgchem.5b00881
-
[13]
Khoenkhoen, N.; de Bruin, B.; Reek, J. N. H.; Dzik, W. I. Eur. J. Inorg. Chem. 2015, 2015, 567.
-
[14]
Yandulov, D. V.; Schrock, R. R. Science 2003, 301, 76.
-
[15]
Arashiba, K.; Miyake, Y.; Nishibayashi, Y. Nat. Chem. 2011, 3, 120. doi: 10.1038/nchem.906
-
[16]
Anderson, J. S.; Rittle, J.; Peters, J. C. Nature 2013, 501, 84. doi: 10.1038/nature12435
-
[17]
(a) Kuriyama, S.; Arashiba, K.; Tanaka, H.; Matsuo, Y.; Nakajima, K.; Yoshizawa, K.; Nishibayashi, Y. Angew. Chem. Int. Ed. 2016, 55, 14291. (b) Hill, P. J.; Doyle, L. R.; Crawford, A. D.; Myers, W. K.; Ashley, A. E. J. Am. Chem. Soc. 2016, 138, 13521.
-
[18]
Shiina, K. J. J. Am. Chem. Soc. 1972, 94, 9266. doi: 10.1021/ja00781a068
-
[19]
Komori, K.; Oshita, H.; Mizobe, Y.; Hidai, M. J. Am. Chem. Soc. 1989, 111, 1939. doi: 10.1021/ja00187a092
-
[20]
Tanaka, H.; Sasada, A.; Kouno, T.; Yuki, M.; Miyake, Y.; Nakanishi, H.; Nishibayashi, Y.; Yoshizawa, K. J. Am. Chem. Soc. 2011, 133, 3498. doi: 10.1021/ja109181n
-
[21]
Yuki, M.; Tanaka, H.; Sasaki, K.; Miyake, Y.; Yoshizawa, K.; Nishibayashi, Y. Nat. Commun. 2012, 3, 1254. doi: 10.1038/ncomms2264
-
[22]
Imayoshi, R.; Tanaka, H.; Matsuo, Y.; Yuki, M.; Nakajima, K.; Yoshizawa, K.; Nishibayashi, Y. Chem. Eur. J. 2015, 21, 8905. doi: 10.1002/chem.201501088
-
[23]
Morello, L.; Yu, P.; Carmichael, C. D.; Patrick, B. O.; Fryzuk, M. D. J. Am. Chem. Soc. 2005, 127, 12796. doi: 10.1021/ja054467r
-
[24]
MacLeod, K. C.; Holland, P. L. Nat. Chem. 2013, 5, 559. doi: 10.1038/nchem.1620
-
[25]
Volpin, M. E.; Shur, V. B.; Kudryavtsev, R. V.; Prodayko, L. A. Chem. Commun. 1968, 1038.
-
[26]
van Tamelen, E. E.; Rudler, H. J. Am. Chem. Soc. 1970, 92, 5253. doi: 10.1021/ja00720a061
-
[27]
Hori, K.; Mori, M. J. Am. Chem. Soc. 1998, 120, 7651. doi: 10.1021/ja981465g
-
[28]
Mori, M.; Uozumi, Y.; Shibasaki, M. Tetrahedron Lett. 1987, 28, 6187. doi: 10.1016/S0040-4039(00)61842-7
-
[29]
Ueda, K.; Sato, Y.; Mori, M. J. Am. Chem. Soc. 2000, 122, 10722. doi: 10.1021/ja002707r
-
[30]
Knobloch, D. J.; Lobkovsky, E.; Chirik, P. J. Nat. Chem. 2010, 2, 30. doi: 10.1038/nchem.477
-
[31]
Curley, J. J.; Sceats, E. L.; Cummins, C. C. J. Am. Chem. Soc. 2006, 128, 14036. doi: 10.1021/ja066090a
-
[32]
Guru, M. M.; Shima, T.; Hou, Z. Angew. Chem. Int. Ed. 2016, 55, 12316. doi: 10.1002/anie.201607426
-
[33]
Semproni, S. P.; Margulieux, G. W.; Chirik, P. J. Organometallics 2012, 31, 6278. doi: 10.1021/om3005542
-
[34]
Klopsch, I.; Kinauer, M.; Finger, M.; Würtele, C.; Schneider, S. Angew. Chem. Int. Ed. 2016, 55, 4786. doi: 10.1002/anie.201600790
-
[35]
Sellmann, D.; Weiss, W. Angew. Chem. Int. Ed. 1978, 17, 269. doi: 10.1002/(ISSN)1521-3773
-
[36]
Watakabe, A.; Takahashi, T.; Jin, D. M.; Yokotake, I.; Uchida, Y.; Hidai, M. J. Organomet. Chem. 1983, 254, 75. doi: 10.1016/0022-328X(83)85119-5
-
[37]
Bernskoetter, W. H.; Lobkovsky, E.; Chirik, P. J. Angew. Chem. Int. Ed. 2007, 46, 2858. doi: 10.1002/(ISSN)1521-3773
-
[38]
Knobloch, D. J.; Toomey, H. E.; Chirik, P. J. J. Am. Chem. Soc. 2008, 130, 4248. doi: 10.1021/ja8008506
-
[39]
Semproni, S. P.; Chirik, P. J. J. Am. Chem. Soc. 2013, 135, 11373. doi: 10.1021/ja405477m
-
[40]
Keane, A. J.; Farrell, W. S.; Yonke, B. L.; Zavalij, P. Y.; Sita, L. R. Angew. Chem. Int. Ed. 2015, 54, 10220. doi: 10.1002/anie.201502293
-
[41]
Seino, H.; Ishii, Y.; Sasagawa, T.; Hidai, M. J. Am. Chem. Soc. 1995, 117, 12181. doi: 10.1021/ja00154a019
-
[42]
Hori, M.; Mori, M. J. Org. Chem. 1995, 60, 1480. doi: 10.1021/jo00111a001
-
[43]
Amarnath, V.; Anthony, D. C.; Amarnath, K.; Valentine, W. M.; Wetterau, L. A.; Graham, D. G. J. Org. Chem. 1991, 56, 6924. doi: 10.1021/jo00024a040
-
[44]
Mori, M.; Hori, K.; Akashi, M.; Hori, M.; Sato, Y.; Nishida, M. Angew. Chem. Int. Ed. 1998, 37, 636. doi: 10.1002/(ISSN)1521-3773
-
[45]
Pickett, C. J.; Leigh, G. J. Chem. Commun. 1981, 1033.
-
[1]
-

图 6 以芳香卤代烃和氮气为原料制备芳香胺类产物
Figure 6 Synthesis of aromatic amines from aromatic halohydrocarbon and dinitrogen

图 8 以芳香酰氯和氮气为原料制备芳香酰胺及酰亚胺
Figure 8 Synthesis of aromatic amides and imides from aroyl chloride and dinitrogen

图 11 以芳香卤代烃和一氧化碳为原料制备酰胺及酰亚胺
Figure 11 Synthesis of aromatic amides and imides from aromatic halohydrocarbon and carbon monoxide

图 14 以氮气为原料合成有机腈类化合物的反应循环图
Figure 14 A synthetic cycle that incorporates dinitrogen into organic nitriles

图 15 以氮气和酰氯为原料合成腈类化合物
Figure 15 Synthesis of nitrile from dinitrogen and acyl chloride

图 16 以氮气和一氧化碳为原料合成乙腈
Figure 16 Synthesis of acetonitrile from dinitrogen and carbon monoxide

图 17 以氮气为原料合成乙腈的反应循环图
Figure 17 A synthetic cycle that incorporates dinitrogen into acetonitrile

图 18 以氮气为原料合成二氮烯类化合物
Figure 18 A synthetic cycle that incorporates dinitrogen into diazenes

图 20 以氮气和二氧化碳为原料合成连氮类化合物
Figure 20 Synthesis of azine from dinitrogen and carbon dioxide

图 21 以氮气和二氧化碳为原料合成连氮类化合物
Figure 21 Synthesis of azine from dinitrogen and carbon dioxide

图 22 以氮气和一氧化碳为原料合成碳二亚胺类化合物
Figure 22 Synthesis of carbodiimide from dinitrogen and carbon monoxide

图 23 以氮气为原料合成异氰酸酯类化合物的反应循环图
Figure 23 A synthetic cycle that incorporates dinitrogen into isocyanate

图 24 以氮气为原料合成吡咯和N-氨基吡咯
Figure 24 Synthesis of pyrrole and N-amino pyrrole from dinitrogen

图 25 以氮气为原料合成吡咯和N-氨基吡咯的催化循环过程
Figure 25 A synthetic cycle that incorporates N2 into pyrrole and N-amino pyrrole

图 26 以氮气为原料合成β位有取代基的吡咯
Figure 26 Synthesis of pyrrole with a substituent on the β position from dinitrogen

图 27 以氮气为原料合成吲哚及喹啉类衍生物
Figure 27 Synthesis of derivatives of indole and quinoline from dinitrogen

图 29 以氮气为原料合成吡呤类及吲嗪类衍生物
Figure 29 Synthesis of derivatives of pyrrolizine and indolizine from dinitrogen
-


 下载:
下载:





























 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 113
- 文章访问数: 6332
- HTML全文浏览量: 1520
 下载:
下载:
