最近10年,材料晶体结构预测技术取得了重大突破[1 3 。晶体结构预测技术可能使新材料发现的周期会大大缩短,也成为材料基因组的重要技术之一[4 6 。例如,通过晶体结构预测,研究人员已经发现了一系列新型的超硬材料[7 9 、超导材料[10 11 、拓扑绝缘体[12 等。除了辅助用于新材料的发现外,晶体结构预测技术的另一应用是研究人员能够通过无偏见的理论预测来获得任意给定材料体系的潜在晶体结构[1 。这一重要应用有助于消除研究人员对某些材料晶体结构的不确定性,并进而对材料的合成和使用提供指导。近几年来,研究人员运用晶体结构理论预测技术针对过渡金属硼化物体系进行了很多研究[13 16 。一方面,是因为过渡金属硼化物往往兼具十分优异的力学、热学和电学等性能,是一类应用广泛和具有重大研究价值的材料[17 18 ;另一方面,则是因为过渡金属硼化物往往能够形成多种配比的稳定晶体结构,而仅依靠于实验研究并不能十分完全地发现和确定它们的晶体结构。晶体结构决定材料性质。因此,通过理论预测出不同配比过渡金属硼化物的晶体结构并进而研究它们的理论性质,无疑将对过渡金属硼化物的实验合成和实际应用起到十分重要的指导作用。
本研究采用晶体结构预测技术确定过渡金属硼化铪体系(HfB)的晶体结构。根据实验和理论研究报道[19 ,硼化铪具有2种不同的化学计量比形式,一硼化铪(HfB)和二硼化铪(HfB2 )。HfB2 是一种常用的超高温陶瓷材料,具有十分优异的材料综合性能如高熔点、高硬度、高力学强度等[20 。HfB2 具有AlB2 型晶体结构,属六方晶系结构,空间群为P 6/mmm 。相比于HfB2 ,HfB的确切晶体结构和性质仍存在诸多疑问。研究人员仅仅猜测HfB可能具有FeB型[19 、CrB型[21 、MoB型[22 或NaCl型[23 晶体结构。基于这些HfB晶体结构进行的理论计算表明,FeB型HfB晶体结构的基态能量最低,是基态下最稳定的晶体结构[21 23 。然而,考虑到迄今为止并无直接证据表明HfB具有FeB型晶体结构,因而有必要利用晶体结构预测技术针对HfB的晶体结构进行全局搜索。特别要指出的是,研究人员通过晶体结构预测已成功纠正过很多过渡金属硼化物的晶体结构。在本研究中,通过晶体结构预测,发现了2个比FeB型结构具有更低基态能量的HfB晶体结构。通过第一性原理计算,系统地论证了这2个新发现结构的晶格动力学和力学稳定性。进一步地,基于新发现的2个和已报道的4个HfB晶体结构,运用密度泛函微扰理论研究了温度对HfB晶体结构稳定性的影响。作为补充,还通过第一性原理计算给出了这些HfB材料的力学性质和电子性质。研究结果可为HfB材料的后续实验和理论研究提供依据。
1.
计算方法
采用基于进化算法开发的晶体结构预测软件USPEX[1 2 对HfB的晶体结构进行全局搜索。仅需指定化学元素种类、原子个数和目标性质等基本参数信息,并结合外部接口软件对目标性质进行计算,USPEX软件便可以利用遗传和变异等进化操作手段发现该指定化学体系下具有预期优选性质的晶体结构。当用户选择能量最低作为优化条件时,USPEX软件搜索得到的便是最稳定的晶体结构。在当前的晶体结构预测中,考虑每一晶胞可包含1~4倍化学计量比的HfB。在密度泛函理论(DFT)[24 的框架下,针对晶体结构预测过程中由USPEX软件产生的每一晶体结构,运用VASP软件包[25 对其进行充分的几何结构优化和总能计算,具体参数设置如下:采用全电子投影缀加波(PAW)[26 方法,广义梯度近似(GGA)Perdew-Burke-Emzerhof(PBE)[27 形式,平面波截断能设置为500 eV,倒易空间k 点网格分辨率为2π×0.6 nm-1 。以上这些参数设置既可以保证晶体结构的能量得到较好收敛又能确保USPEX软件进行结构预测的准确性和效率。对于预测发现的2个新型低能量HfB晶体结构,连同文献已报道的4个晶体结构一起,利用VASP软件并采用更小的倒易空间k 点网格分辨率(2π×0.3 nm-1 )对它们的结构、力学和电子结构等性质进行计算。需要说明,考虑到某些HfB晶体结构能量间的微小差异,对以上相关计算参数的选取已进行了严格和全面的测试,测试结果表明选用的计算参数是合理的。为了验证新发现的2个HfB晶体结构的晶格动力学稳定性,采用Phonopy软件[28 基于密度泛函微扰理论(DFPT)[29 计算了这些晶体结构的声子色散曲线。此外,采用VESTA软件[30 对HfB晶体结构进行可视化展示。
2.
研究结果
2.1
HfB晶体结构的预测
为了探究HfB的确切晶体结构,研究人员已经提出过4种可能的晶体结构类型[19 21 23 :FeB型(空间群:Pnma )、CrB型(空间群:Cmcm )、MoB型(空间群:I 41 /amd )、以及NaCl型(Fm 3 m )。基于这4个HfB晶体结构进行的理论研究表明:Pnma 结构具有最低的基态能量,因而是基态下最稳定的HfB晶体结构。通过对HfB的基态晶体结构进行全面预测,发现了2个比Pnma 结构具有更低基态能量的晶体结构:P 6 m 2和R 3 m 。在不考虑零点能的情况下,相比于Pnma 结构,P 6 m 2结构的平均每原子能量相比Pnma 结构要低12 meV;R 3 m 结构的能量略低于Pnma 结构,二者能量相差不到2 meV·atom-1 。
对新发现的2个以及已报道的4个HfB晶体结构进行了充分的几何结构优化,这些晶体结构优化后的几何结构信息列于表 1 表 1 [23 。通过对比发现,本次计算结果与前人的报道结果基本一致,表明了当前计算结果的正确性和可靠性。表 1 P 6 m 2结构的能量最低,R 3 m 结构的能量次低,然后依次是Pnma 结构,Cmcm 结构,I 41 /amd 结构和Fm 3 m 结构。如果仅从能量角度考虑不同结构间的稳定性差异,以上6个HfB晶体结构在基态下的稳定性由高到低的次序为:P 6 m 2→R 3 m →Pnma →Cmcm →I 41 /amd →Fm 3 m 。需要指出的是,前4个HfB晶体结构(P 6 m 2,R 3 m ,Pnma 和Cmcm 结构)间的能量差异较小(≤20 meV·atom-1 ),意味着这4个HfB晶体结构实验存在的可能性较大并需要被重点关注。为了便于后续的实验验证,给出了这6个HfB晶体结构的模拟X射线衍射图,如图 1
表 1
图 1
2.2
HfB晶体结构的稳定性
理论上,判断一个晶体结构在基态下的稳定性,除了需要考虑晶体结构的基态能量外,还需要考虑晶体结构的晶格动力学和力学稳定性。晶体结构的晶格动力学稳定性可以由其声子色散曲线反映出来,力学稳定性则可以通过验证其弹性常数是否符合Born-Huang稳定准则[31 进行判断。
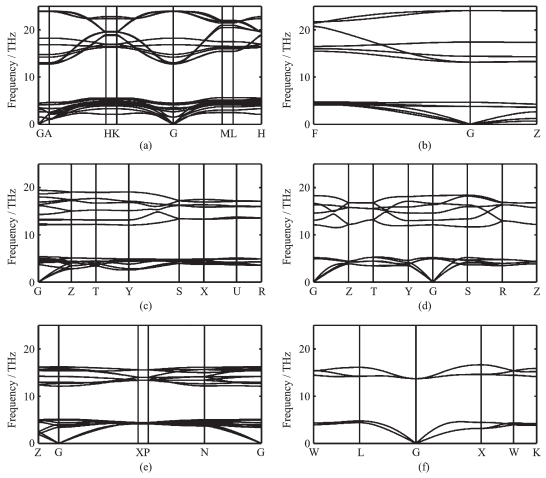
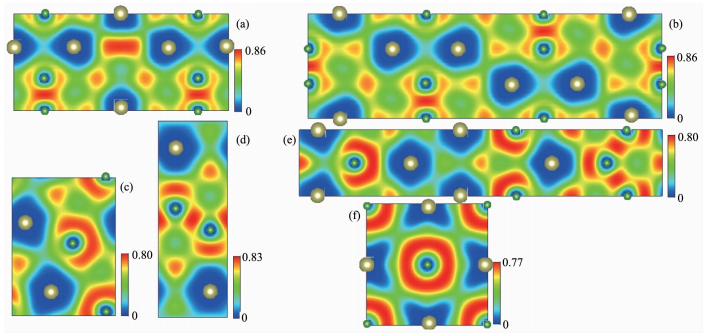
对于新发现的P 6 m 2和R 3 m 结构,首先分别构建了3×3×1和2×3×3超胞结构,然后利用这2个超胞结构计算了它们的声子色散曲线。如图 2(a) (b) P 6 m 2和R 3 m 结构的声子振动频率均无虚频,说明这2个新发现的HfB晶体结构在基态下均具有晶格动力学稳定性。本研究也补充验证了已报道的4个HfB晶体结构在基态下的晶格动力学稳定性,如图 2(c) (f)
图 2
针对以上6个HfB晶体结构,进一步计算了它们的弹性常数并依据Born-Huang稳定准则对它们的力学稳定性进行了判断。通过判断,这6个HfB晶体结构在基态下均具有力学稳定性。通过计算获得的HfB晶体结构的独立弹性常数列于表 2
表 2
2.3
温度对HfB晶体结构能量的影响
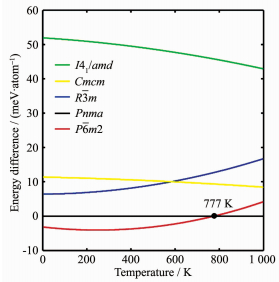
上文中已经比较了不同HfB晶体结构在基态下的能量。这里进一步考虑温度对HfB晶体结构能量的影响。当不考虑非谐效应时,晶体结构在零压强以及不同温度条件下的自由能可通过如下公式进行计算: $F\left( T \right) = {E_{{\rm{tot}}}} + {E_{{\rm{zpe}}}} + kT\int {f\left( \omega \right){\rm{ln}}\left[ {1 - {\rm{exp}}\left( {\frac{{h\omega }}{{kT}}} \right)} \right]{\rm{d}}\omega } $ ;式中F (T )即晶体结构在不同温度T 下的自由能,E tot 是晶体结构在基态下的能量(表 1 E zpe 是晶体结构的零点能( ${E_{{\rm{zpe}}}} = \int {f\left( \omega \right)h\omega {\rm{d}}\omega } $ ),k 为玻尔兹曼常数,h 为普朗克常数,f (ω )是声子态密度, ω 是声子振动频率。基于密度泛函微扰理论,通过计算得到了6个HfB晶体结构的声子态密度,并由此获得了这些HfB晶体结构在不同温度下的自由能,如表 3 表 1 表 3 R 3 m 结构在0 K时的能量会高于已报道的Pnma 结构,而其他结构的能量大小顺序没有改变。零点能修正的结果表明:在不考虑零点能的情况下进行晶体结构预测时,研究人员不能仅仅只关注能量最低的结构,还需要关注其他一些能量较低的结构。
表 3
利用表 3 Pnma 结构的自由能作为零基准,进一步获得了其它5个HfB晶体结构的自由能随温度的变化曲线,如图 3 Fm 3 m 结构与Pnma 结构的能量相差过大(> 200 meV· atom-1 ),因此图 3 Fm 3 m 结构的信息。不难发现,温度可以显著改变不同HfB晶体结构间的能量差异甚至是相对大小。例如,在0 K下,新发现的P 6 m 2结构比Pnma 结构具有更低的能量。但是,当T > 777 K后,Pnma 结构的能量却比P 6 m 2结构的能量低。类似的现象同样可见于R 3 m 结构和Cmcm 结构:当T < 585 K时,R 3 m 结构的能量比Cmcm 结构的能量小;而当T > 585 K时,Cmcm 结构比R 3 m 结构具有更低的能量。
图 3
由图 3 P 6 m 2、R 3 m 、Pnma 和Cmcm 这4个HfB晶体结构间的能量大小次序发生了变化,但是这些结构间的能量差异却始终维持在20 meV·atom-1 以内。这说明,即便在高温条件下,这4个HfB晶体结构也均有较大可能被实验合成和发现。而对于I 41 /amd 和Fm 3 m 结构来说,由于它们具有相对较高能量,推测它们实际存在的可能性要低得多。
2.4
HfB晶体结构的结构特征
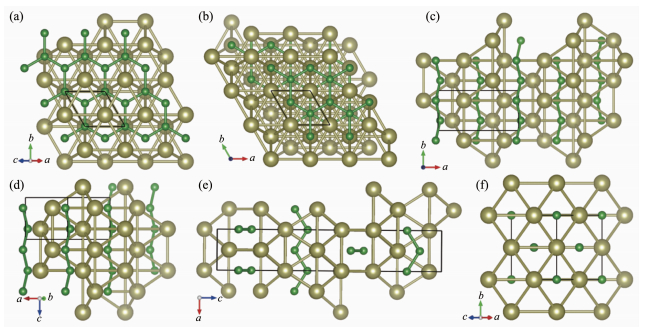
图 4 图 4(a) (b) P 6 m 2和R 3 m 结构中,每个B原子与6个Hf原子相连形成Hf6 B正三棱柱,每2个Hf6 B三棱柱通过共面形成Hf8 B2 四棱柱,B原子相互连接形成二维类石墨烯B层,B-B键长约为0.182 nm。如图 4(c) (e) Pnma 、Cmcm 和I 41 /amd 结构中,每个B原子与6个Hf原子相连形成Hf6 B三棱柱(近正三棱柱,略有扭曲),每2个Hf6 B三棱柱通过共面形成Hf8 B2 四棱柱(近棱形四棱柱,略有扭曲),B原子相互连接形成zig-zag链而不组成二维类石墨烯B层,B-B键长范围为0.189~0.199 nm。在Fm 3 m 结构中,每个B原子与6个Hf原子相连形成Hf6 B正八面体,B原子之间不相邻,如图 4(f)
图 4
依据B原子的分布形式,将这6个HfB晶体结构大致分为3大类;第Ⅰ类:含二维类石墨烯B层结构(P 6 m 2和R 3 m 结构);第Ⅱ类:含zig-zag形B链结构(Pnma 、Cmcm 和I 41 /amd 结构);第Ⅲ类:孤立B原子结构(Fm 3 m 结构)。
因为B原子的分布不同,这3种不同类型的HfB晶体结构的性质将大不一样。例如,这3类HfB晶体结构的基态能量有区别(见第2.1小节)。第Ⅰ类HfB晶体结构的基态能量最低;第Ⅱ类HfB晶体结构的基态能量稍高;第Ⅲ类HfB晶体结构的基态能量最高。下面将进一步描述这3类HfB晶体结构力学性质和电子结构的差异。
2.5
HfB的力学性质
HfB材料的一大应用是作为结构陶瓷材料,因而关注它的力学性质是十分必要的。利用计算的弹性常数,基于Voigt-Reuss-Hill近似[32 34 首先估算了多晶HfB材料的体模量B 和剪切模量G 。基于估算的体模量B 和剪切模量G ,进一步获得了多晶HfB材料的杨氏模量E 、泊松比ν 、Pugh比k 以及维氏硬度H v 等。相关计算公式为[35 36 :E =9BG /(3B +G );ν =0.5(3B -2G )/(3B +G );k =G /B ;H v =2(G 3 /B 2 )0.585-3。
表 4 表 4 [37 :若Pugh比大于0.57,材料为脆性材料,反之则为韧性材料。如表 4 [38 :典型共价材料的泊松比要小于0.1,而金属材料的泊松比一般大于0.33。以上3类HfB材料泊松比的相对大小次序为:第Ⅱ类 < 第Ⅰ类 < 第Ⅲ类,表明第Ⅱ类HfB材料中共价键的方向性最强,因而力学强度也最高。
表 4
2.6
HfB晶体结构的电子性质
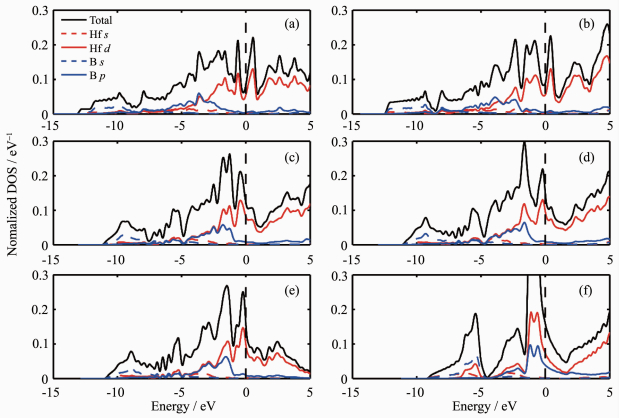
材料的电子结构能够帮助研究人员更为充分地了解材料的性质。通过理论计算,得到了6个HfB晶体结构的电子态密度(包括总态密度和分波态密度)。如图 5 d 电子。由图 5 d 电子与B的2p 电子的态密度曲线具有类似的形状,意味着Hf的5d 电子轨道与B的2p 电子轨道存在杂化。这说明在这6个HfB晶体结构中可能均存在较强的Hf-B共价键。
图 5
为了进一步了解6个HfB晶体结构中的化学键,计算了它们的电子局域函数(ELF[39 )。ELF取值0~1,其数值大小可用于区分金属键、共价键和离子键[40 :ELF=1表示纯共价键或孤对电子,ELF=0.5则对应均匀的电子气。图 6
图 6
正是由于晶体结构中具有强的B-B和Hf-B的共价键,第Ⅰ类和第Ⅱ类HfB材料具有高的弹性模量和维氏硬度。而由于缺少B-B共价键,相比于第Ⅰ类和第Ⅱ类HfB材料来说,第Ⅲ类HfB材料弹性模量和维氏硬度明显偏低。
3.
结论
利用晶体结构预测软件USPEX,系统搜索了HfB在基态下的稳定晶体结构。新发现2个HfB晶体结构(空间群:P 6 m 2和R 3 m )。相比于文献已报道的4个HfB晶体结构(空间群:Pnma 、Cmcm 、I 41 /amd 和Fm 3 m ),P 6 m 2结构具有最低的基态能量。计算了这6个HfB晶体结构在0~1 000 K温度范围内的自由能。计算结果表明:温度能够改变不同HfB晶体结构间能量的大小次序。特别地,当T > 777 K时,Pnma 结构会取代P 6 m 2结构成为自由能最低的HfB晶体结构。这是由于HfB晶体结构中B原子连接方式的差异性所引起的。在P 6 m 2结构中,B原子间相互连接形成的二维类石墨烯B层造成高的声子振动频率,因而P 6 m 2结构的自由能随温度升高而增加的量更多。而在Pnma 结构中,B原子仅通过相互连接形成zig-zag形B链,因而Pnma 结构的声子振动频率较低,进而造成其自由能随温度升高而增加的量相对更少。基于6个不同的HfB晶体结构,计算了它们的力学性质。除具有Fm 3 m 结构的HfB材料外,其它5种HfB材料均具有较好的力学强度。考虑到Fm 3 m 结构稳定存在的可能性较低,预测实际的HfB材料极有可能具有良好的力学强度。进一步的电子结构分析表明:造成HfB材料具有良好力学强度的原因是由于其晶体结构中具有强的B-B和Hf-B的共价键。本研究是材料基因组技术用于材料发现与功能性质基因分析的典型案例,对加速新型功能材料的研发与应用具有重要的借鉴价值。

 下载:
下载:


 下载:
下载:







 下载:
下载:
