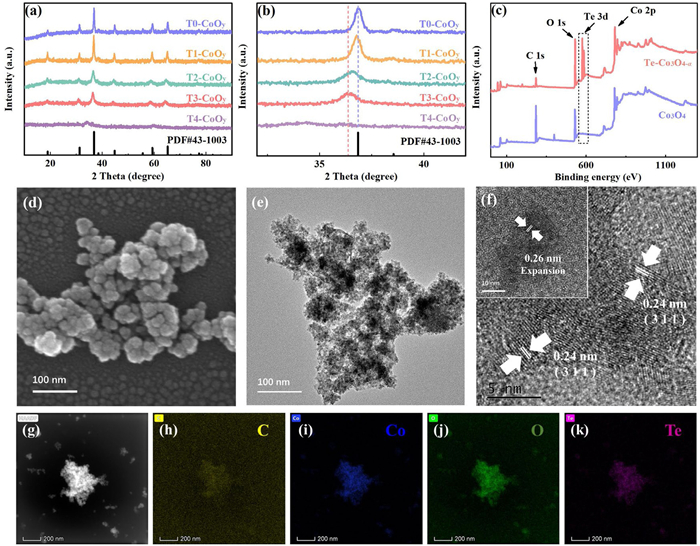
Figure 1.
(a) XRD patterns of prepared samples, (b) locally amplified XRD patterns, (c) total XPS spectra, (d) SEM image, (e) TEM image, (f) HR-TEM image and (g-k) elements mapping of Te-Co3O4-α.

Advanced oxidation processes (AOPs) [1–4], particular those based on heterogeneous activation of peroxymonosulfate (PMS) represent a cutting-edge technology for effectively removing refractory organic contaminants, demonstrating significant potential in water purification applications [4]. The design and development of effectual and steady heterogeneous catalysts are crucial for advancing this technology. Transition metal ions and their oxides, characterized by the versatile valence electron structures and cost-effectiveness, have been extensively utilized in catalyst development. In particular, Co3O4 with a spinel structure has received significant attention [5,6]. However, despite the notable PMS activation capabilities of transition metal oxides, their limited atomic utilization efficiency and the restricted regeneration capacity of low-valent metals have hindered their broader application. Heteroatom doping modification has emerged as one of ideal strategies to elevate the catalytic activity of metal oxide materials [7]. It primarily involves incorporating different atoms into the host oxides via various doping methods (such as atomic substitution, interstitial insertion, and surface adsorption) to optimize pore structure regulation, vacancy defect construction, and electronic structure reconstruction without substantially altering the fundamental crystal structure [8–10]. These modifications to the original chemical environment subsequently influence the interfacial interactions within the reaction system, thereby enhancing catalytic degradation efficiency.
While significant progress has been made in advanced oxidation research through metal oxide doping engineering, the precise link between specific doping types and PMS activation pathways has not been well unlocked and remains inadequately explored. For instance, when doping induces substantial changes in the crystal phase (transitioning from one crystal structure to another or multiple phases), the classification of such phase-transforming processes as doping warrants reconsideration [11,12]. Comparative analysis of activation mechanisms between such "doping" systems and original metal oxides may obscure our understanding of the mechanism underlying enhanced catalytic activity through doping strategies. What is more, different doping types correspond to distinct structural active centers, which not only affect the physicochemical properties of the catalyst but also directly influence the optimization direction of PMS activation progress and types of reactive oxygen species (ROS) generated [13–15]. Less than satisfactory, most studies tend to thoughtlessly attribute doping modalities to a single type for discussion with same dopant [8,9,16]. The research community appears to unintentionally overlook the potential synergistic effects and contributions that may emerge from the coexistence of multiple doping modalities. This excessive conceptual generalization, along with insufficient consideration of coexisting doping modalities, may lead to misconceptions about ROS generation mechanisms and hinder the development of more efficient advanced oxidation systems. Thus, the "one-to-one" structure-activity relationship between doping modalities and activation behaviors merits deep study.
Oxygen vacancy (OV), as an intrinsic defect in metal oxides, has been widely recognized for its significance in activation reaction processes [17]. The doping process in metal oxides frequently generates additional OV via charge compensation mechanisms. However, the specific mechanism by which they influence catalytic activity remains a subject of debate. Numerous studies have demonstrated a positive correlation between OV content and pollutant degradation efficiency [18,19]. However, other research suggests that the coordination environment, rather than simple concentration of OV, is the critical factor determining the intrinsic catalytic activity of the catalyst [20]. In some studies, the OV is considered as the primary catalytically active center to directly enhance ROS production [21,22]. However, direct evidence supporting OV as active sites remains lacking, and this perspective may not adequately explain the various OV-driven systems. Currently, the precise contribution mechanism of OV in PMS activation processes remains unclear. The prevailing notion that OV simultaneously serves as molecular adsorption sites, electron acceleration sites, and active catalytic sites may be an oversimplification and potentially misleading. These uncertainties pose a significant challenge in achieving precise control over the application of OV. Thus, constructing a catalytic degradation system with a well-defined OV action mechanism facilitates a deeper understanding of OV. Also, investigating the correlation between doping types and PMS activation pathways is crucial for advancing the study of catalytic activity in metal oxides based on non-phase transition doping.
To address above issues, here, via a one-step hydrothermal strategy, we reported the synthesis of Te-doped Co3O4 catalyst (i.e., Te-Co3O4-α), which was characterized by various analytical technologies to reveal its morphology, composition and microstructures. Then, its catalytic capability was evaluated. Our study testified that the incorporation of Te via both interstitial and substitutional doping synergistically enhanced the catalytic activity of Co3O4 toward PMS. Quenching and scavenging trials identified the main active sites and the dominant ROS during the catalytic reaction. Different ROS generation mechanisms resulting from various doping configurations, along with the singular synergetic accelerating effect of OV, were proposed. Finally, potential degradation pathways of levofloxacin (LEV) were discussed through liquid chromatography-mass spectrometry (LC-MS) analysis, and the practical utilization performance of the Te-Co3O4-α/PMS system was evaluated.
As shown in X-ray diffraction (XRD) outcomes in Fig. 1a, the prepared samples, including T0-CoOy (i.e., Co3O4), T1-CoOy, T2-CoOy, T3-CoOy (i.e., Te-Co3O4-α) and T4-CoOy (see synthesis details in Supporting information), exhibit a well-defined crystalline structure. These phases align well with the standard card PDF #43–1003 of Co3O4, indicating that a moderate Te doping level does not alter the crystalline phase structure of the samples. However, the intensities of the diffraction peaks gradually weaken and the peaks broaden as the Te doping amount increases, suggesting a reduction in the crystallinity and a decrease in nanoparticle's size. Notably, the diffraction angle of the main peak exhibits a slight negative shift, which becomes more pronounced as Te doping increases (Fig. 1b). This phenomenon is attributed to lattice expansion induced by the interstitial doping of Te species [23]. As the doping amount further increases, a significant deformation in the crystalline phase of T4-CoOy is observed. The diffraction peaks almost completely disappear, indicating that excessive Te doping has thoroughly disrupted the crystalline phase structure of the samples.
As shown in X-ray photoelectron spectrometer (XPS) spectra in Fig. 1c, the Te 3d spin-orbit splitting peaks appear in the Te-Co3O4-α (i.e., T3-CoOy), with an intensity ratio of 3:2 (Fig. S1a in Supporting information). This confirms that Te is successfully incorporated into the sample. In the high-resolution XPS spectrum, the peak of Te 3d5/2 is located at 576.3 eV, and the double peak spacing between Te 3d5/2 and Te 3d3/2 is 10.5 eV (Fig. S1a), indicating that Te predominantly exists in the form of oxide [24]. The high-resolution XPS spectrum of Co 2p for T0-CoOy exhibits the typical peak pattern of Co3O4 (Fig. S1b in Supporting information). The splitting peaks of Co 2p3/2 and Co 2p1/2 appear at ~780 eV and ~796 eV, respectively, and the Co species with two oxidation states of Co3+ and Co2+ are further fitted. After Te introduction, the Co satellite peaks of Te-Co3O4-α associated with Co-O become more prominent. The relative concentration of Co3+ decreases, while that of Co2+ increases, and the Co3+/Co2+ ratio decreases from 0.68 to 0.53 (Table S1 in Supporting information). This may be due to the formation of lattice defects via Te doing, such as oxygen vacancies, resulting in the reduction of some metal ions to lower states. Notably, the binding energy of the Co 2p spectrum of Te-Co3O4-α shows a significant positive shift. The alteration in the electronic environment surrounding the metal atom results from lattice reconfiguration induced by substitutional doping. This positive shift is a strong signal of a decrease in electron density around the Co site [25]. As displayed in Fig. S1c (Supporting information), in the high-resolution XPS spectrum of O 1s, the peaks can be divided into three components corresponding to lattice oxygen (OL), OV, and hydroxyl oxygen/surface-adsorbed oxygen species (OH) from low to high binding energy [26], respectively. Due to the slightly higher electronegativity of Te compared to Co, the electron density around the O atoms decreases when Co3+ is substituted. Consequently, the peak corresponding to OL shifts from 529.68 eV to a higher binding energy of 530.48 eV. The significant reduction in its relative concentration implies the substantial formation of OV (Table S2 in Supporting information). However, the doping of Te leads to an insignificant increase in the relative content of surface OV. This may be because the Co3O4 synthesized hydrothermally already contains some water molecules, so the peak around 531.4 eV also includes oxygen from adsorbed water. In fact, the subsequent solid electron paramagnetic resonance (EPR) trials (see later discussion) confirmed that Te incorporation significantly increased the content of OV, which further contributed to the improvement of the surface electronic structure. The enhanced surface activity results in a noticeable increase in the relative proportion of adsorbed molecular oxygen species, which also indirectly confirms the increase in OV content [27].
Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) were employed to analyze the morphological structure of the samples and shown in Figs. 1d and e, respectively. The synthesized Co3O4 nanoparticles exhibit a cubic morphology, with an average particle size ranging from 50 nm to 100 nm (Figs. S2a and b in Supporting information). Upon Te incorporation, as shown in Fig. 1d and Fig. S2c (Supporting information), the nanoparticles almost lost their cubic shape, and the particle size reduces to 20–50 nm. This phenomenon is attributed to the doping-induced suppression of crystallization in the sample. In the high-resolution (HR)-TEM, lattice fringes corresponding to the (311) crystal plane of Co3O4 were observed, with a lattice spacing of 0.24 nm (Fig. 1f), which is consistent with the XRD analysis. Simultaneously, a large number of lattice fringes with a spacing of 0.26 nm are noted (inset of Fig. 1f). This abnormal expansion is due to the incorporation of Te, which has a larger atomic radius, resulting in a wider lattice spacing. This strongly indicates the coexistence of interstitial Te and substituted Te [13]. Figs. 1g-k show the elemental mapping, which further confirms the successful doping and uniform distribution of Te in the sample. Overall, the above analysis confirms that the Te-Co3O4-α catalyst maintains the Co3O4 phase with successful Te doping. The higher-valent Te likely substitutes for Co3+ in the octahedral sites, leading to partial reduction of Co species to Co2+. The local Co-O-Te structure is formed and the charge balance is regulated by the formation of oxygen vacancies. Simultaneously, Te also exists in the lattice as an interstitial ion (e.g., TeOx and Te4+), occupying otherwise vacant interstitial sites, which is typically accompanied by lattice volume expansion [23].
The pore structure characteristics of the samples were investigated through Brunauer–Emmett–Teller (BET) analysis. As depicted in Fig. S3a (Supporting information), the N2 adsorption-desorption isotherms of both Co3O4 and Te-Co3O4-α exhibit Type Ⅳ isotherms with H1 hysteresis loops, indicating cylindrical-like pore characteristics. The specific surface areas are 20.29 m2/g for Co3O4 and 126.85 m2/g for Te-Co3O4-α. The successful Te doping appears to further amplify these structural defects, increasing the specific surface area by over 6-fold. This structural change significantly increases the exposure of active sites, greatly facilitates the diffusion of target molecules and the transfer of intermediates, thereby enhancing their catalytic performance. Raman spectroscopy was employed to further analyze the effect of Te doping on the strength of Co-O bond. As shown in Fig. S3b (Supporting information), the Raman peaks of the original Co3O4 at 192, 480, 520, 618 and 687 cm−1 correspond to the tensile vibrations of F2g1, Eg, F2g2, F2g3 and A1g modes [9], respectively. After Te doping modification, Raman peaks exhibit a slight blue shift, and the signal weakens as the peak width increases. This is due to Te doping breaking the local symmetry associated with the oxygen atom, which weakens the Co-O bond strength. Additionally, this change may also be related to quantum size effects or surface effects caused by the reduction in particle size. Oxygen vacancies in the samples are investigated and detected using solid-state EPR spectroscopy. A distinct signal peak is observed in the Te-Co3O4-α at g = 2.004, with an intensity of ~4–5 times that of Co3O4 (Fig. S3c in Supporting information). This provides direct evidence that Te doping induces the formation of rich oxygen vacancies in Co3O4, in good agreement with the aforementioned results.
To evaluate the catalytic activity of samples, an advanced oxidation degradation system based on PMS heterogeneous activation was constructed, targeting LEV as the pollutant of interest (see UV–vis spectrum in Fig. S3d in Supporting information). As shown in Fig. 2a, PMS alone exhibits negligible degradation of LEV. The adsorption kinetics of Te-Co3O4-α is slightly faster than that of Co3O4. This can be attributed to the structural defect reorganization induced by Te doping, which increases the specific surface area as the doping amount rises. This confirms that the catalyst surface provides more pores and active sites, facilitating the adsorption of target molecules. However, the removal rates of LEV for both Te-Co3O4-α and Co3O4 are below 10.0% in 40 min. When both the catalyst and PMS are present simultaneously, the concentration of LEV in the Te-Co3O4-α/PMS system rapidly decreases, while that in the Co3O4/PMS system declines relatively slow (Fig. 2a). This indicates that the doping of Te enhances the PMS activation ability of Co3O4 to generate highly oxidative active species for rapidly decomposing LEV. The calculation and comparison of the apparent rate constant (kapp) for LEV reveal that the Te-Co3O4-α/PMS system (0.2129 min−1) exhibits a 66-fold enhancement compared to the Co3O4/PMS system (0.0032 min−1). Further control trials were carried out to investigate the effect of Te doping on catalytic activity. When using the T0-CoOy, T1-CoOy, T2-CoOy, or T3-CoOy as catalyst, the degradation efficiency of LEV progressively increases under the identical reaction conditions, with LEV removal rates of 13.3%, 80.6%, 91.1%, and 96.5% (Fig. 2b), respectively. The corresponding kapp values are 0.0032, 0.0428, 0.0995, and 0.2129 min−1, respectively. This suggests that the gradual increase in Te doping in Co3O4 enhances the catalyst's activation of PMS. However, when the doping amount was further increased, there was no significant enhancement in the removal rate and degradation kinetics in T4-CoOy/PMS system (95.5% with 0.2129 min−1). Furthermore, the crystal structure of T4-CoOy has been altered due to excessive Te doping (Fig. 1b). The Co leaching content in the reaction solution was determined by flame atomic absorption spectrometry. The concentration of Co ions leached in the T4-CoOy/PMS system (4.13 mg/L) is significantly higher than that in the T3-CoOy/PMS system (0.68 mg/L). Consequently, the T3-CoOy, i.e., Te-Co3O4-α, was selected for the subsequent trials.
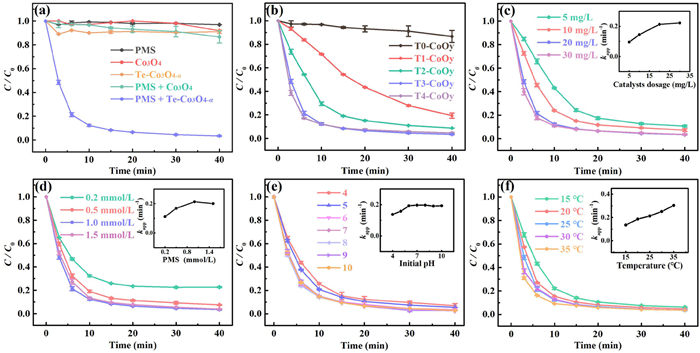
The LEV degradation was affected by various parameters, which were optimized to achieve high LEV degradation performance. As observed in Fig. 2c, the increase in catalyst dosage is positively correlated with the degradation kinetics of LEV. When the catalyst concentration was increased from 5 mg/L to 20 mg/L, the kapp value rose from 0.0959 min−1 to 0.2129 min−1, and LEV removal increased from 90.2% to 96.5%. This is attributed to the increase in catalytic active spots available for reaction, providing more contact of catalyst with substrate. However, further increasing the catalyst's dosage to 30 mg/L results in negligible changes in the removal rate and degradation kinetics (96.4% with 0.2219 min−1). Thus, 20 mg/L of Te-Co3O4-α was selected. Additionally, increasing the PMS concentration from 0.2 mmol/L to 1.0 mmol/L enhances the removal of LEV from 77.3% to 96.5%, with kapp value increasing from 0.1118 min−1 to 0.2129 min−1 (Fig. 2d). This indicates that the predominant active species originate from PMS, and insufficient oxidant can lead to incomplete LEV degradation. However, at 1.5 mmol/L PMS, the LEV removal shows no significant change (96.1%), with a slight kapp decrease (0.2005 min−1). This may be attributed to the self-quenching effect of reactive radicals in a short time (Eqs. 1 and 2) [28]. Moreover, excess PMS can react with radicals, consuming SO4•− and generating the less reactive SO5•− species (Eq. 3) [29]. We further investigated the effect of the initial solution pH. As traced in Fig. 2e, the Te-Co3O4-α/PMS system maintains high LEV removal efficiency over a broad pH range of 4.0-10.0. Notably, when the initial solution is weakly acidic (pH 4.0–5.0), the LEV degradation kinetics is slightly slow. Zeta potential results indicate that the catalyst surface remains negative within the pH from 4.0 to 10.0, ruling out electrostatic interactions as the cause (Fig. S4a in Supporting information). The observed effect may be attributed to the excessive H+ ions, which have a scavenging effect on the generated SO4•− and •OH (Eqs. 4 and 5) [30]. The reduction in active radicals consequently leads to a declined LEV degradation. Reaction temperature is also an important factor controlling LEV degradation. As displayed in Fig. 2f, increasing temperature leads to an upward trend in the degradation kinetics of LEV, i.e., the kapp increases from 0.1350 min−1 to 0.3039 min−1 with temperature from 15 ℃ to 35 ℃. This is because higher temperature can enhance the generation rate of radicals, and increase the collision frequency between active species and substrate molecules, and thus speed up the reaction rate. Finally, 25 ℃ was selected in our work. As shown in Fig. S4b (Supporting information), the linear relationship between lnkapp and 1/T follows the Arrhenius equation (Eq. S2 in Supporting information), with a correlation coefficient of 0.9719. The estimated activation energy (Ea) is 28.39 KJ/mol (see details in Supporting information). This low activation energy suggests high activation efficiency of PMS by Te-Co3O4-α. This enables the Te-Co3O4-α/PMS system to operate efficiently, offering potential for practical applications.
|
|
(1) |
|
|
(2) |
|
|
(3) |
|
|
(4) |
|
|
(5) |
In the PMS-based AOPs, common reactive oxygen species include SO4•−, •OH, and O2•−. Methanol exhibits a certain quenching ability towards SO4•−, while tert–butyl alcohol (TBA), which has a high affinity for •OH, can be employed to differentiate the roles of •OH and SO4•− [31]. Additionally, p-benzoquinone (p-BQ) can serve as a specific scavenger for O2•− [32]. The effect of TBA is minimal, with < 5.0% LEV degradation inhibition even at high TBA concentration (Fig. S5a in Supporting information). In contrast, methanol exhibits a relatively higher inhibition effect, with at least 16.8% decline of LEV degradation. Furthermore, LEV degradation inhibition is proportional to methanol concentration (Fig. S5b in Supporting information). This suggests that the contribution of •OH to LEV degradation is minimal and can be neglected, while that of SO4•− is relatively more significant, though the degradable portion is still limited. In the EPR tests, both the characteristic signals of 5,5-dimethyl-1-pyrroline N-oxide (DMPO)-SO4•− and DMPO-•OH products were simultaneously detected (Fig. 3a), confirming that both SO4•− and •OH are generated during the activation process. Moreover, the characteristic signal of the DMPO-O2•− adduct was also captured, indicating the generation of O2•− in the Te-Co3O4-α/PMS system (Fig. 3b). The addition of p-BQ inhibited 32.2% of the LEV removal (Fig. S5c in Supporting information). Thus, it can be inferred that O2•− plays a non-negligible role in LEV degradation, either by directly oxidizing the target molecule or by converting to 1O2, which further decomposes the compound (Eqs. 11 and 12) [33].
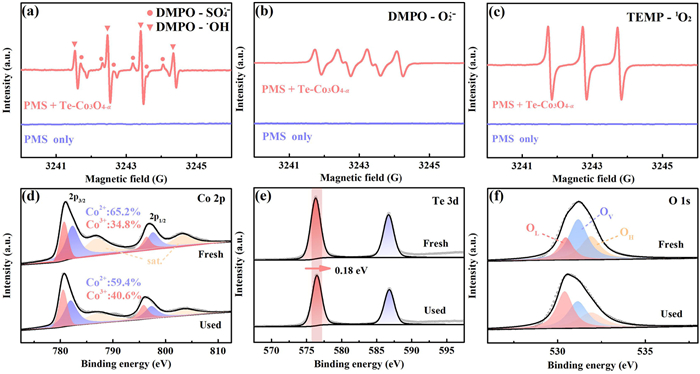
In addition to the radical pathway, non-radical pathways are also common oxidative mechanisms in PMS-based advanced oxidation systems, typically involving the generation of 1O2, the formation of high-valent metal-oxygen (HVMO), and material-mediated electron transfer processes (ETP) [34]. L-Histidine has been used in previous studies to quench 1O2 and assess its contribution [35]. As shown in Fig. S5d (Supporting information), the addition of L-histidine significantly inhibited the degradation process, with the inhibition ratio up to 74.6%. This implies 1O2 may be formed and participates in LEV removal, verified by the EPR trial (Fig. 3c). Based on this, it is inferred that 1O2 may also be one of the ROS responsible for the LEV degradation. The Cr2O72− was used as an electron scavenger to investigate the contribution of the ETP [36]. As displayed in Fig. S5e (Supporting information), upon the addition of K2Cr2O7 to the system, LEV degradation was inhibited by 42.8%. To confirm whether the ETP indeed occurs, we pre-mixed the catalyst and PMS to construct and accumulate the surface-bound PMS complex (catalyst-PMS*), and found that a pre-mixing of 10 min had almost no impact on the degradation kinetics (Fig. S6a in Supporting information). This result suggests that the ETP's contribution may be controversial. As shown in Fig. S6b (Supporting information), the i-t curve displays the current variation of Te-Co3O4-α at open-circuit voltage. When PMS was added at 60 s, a distinct current peak appeared immediately, originating from the rapid interaction between Te-Co3O4-α and PMS. After the signal stabilized, the injected LEV caused almost no further current signal changes, indicating that no catalyst-PMS* was involved in triggering the ETP with LEV. Therefore, the inhibitory effect of K2Cr2O7 may be caused by the depletion of the catalytic site [15]. HVMO, produced through the oxygen transfer reaction between metal sites and PMS, represents a typical non-radical oxidation pathway. Dimethyl sulfoxide (DMSO) can be used as a chemical probe to evaluate the contribution of HVMO species [37]. The addition of DMSO significantly inhibited the degradation kinetics of LEV (Fig. S7a in Supporting information). This suggests that HVMO, i.e., Co(Ⅳ)=O, may be generated in the Te-Co3O4-α/PMS system and plays a crucial role in the degradation of LEV. DMSO can act as a scavenger for surface-bound radicals [38]. Therefore, further investigation was conducted to study the desorption effect of F− on surface-bound radicals and their contribution to LEV degradation [39]. The control results show the minor effect of NaF on the degradation kinetics (Fig. S7b in Supporting information). This indicates that the contribution of surface-bound radicals is negligible, further confirming the critical role of the Co(Ⅳ)=O in LEV degradation. Above outcomes indicate that no ETP occurred in the Te-Co3O4-α/PMS system. The SO4•−, O2•−, 1O2, and Co(Ⅳ)=O all make significant contributions to the LEV removal, while the contribution of •OH is relatively minor.
To elucidate the mechanism of PMS activation by Te-Co3O4-α, further investigation was conducted to trace the primary active sites of the catalyst. KSCN was employed to neutralize the Co sites during the reaction to explore the role of the metal sites [25]. As expected, the addition of KSCN almost completely inhibited LEV degradation (Fig. S8a in Supporting information), demonstrating the irreplaceable core role of the metal sites in activating PMS to generate active spots. The substantial formation of oxygen vacancies induced by Te doping may also be associated with the enhancement of catalytic activity, making it crucial to clarify the role of OV in the activation reaction. Na2S, which can sulfidize the surface via an O2−/S2− anion exchange, was used to analyze the contribution of OV in the activation/degradation process [21]. As displayed in Fig. S8b (Supporting information), after sulfidation treatment on the Te-Co3O4-α, the LEV removal only decreased by 4.2%. However, its degradation kinetics slowed down significantly, and the extent of weakening increased with Na2S' concentration. This indicates that the elimination of OV primarily hinders PMS activation rate by Te-Co3O4-α, implying that OV likely controls the rate-limiting step of PMS activation rather than directly involving the generation of ROS. The positive impact of Te doping and OV formation on electron transfer efficiency was further studied through electrochemical trials. The Nyquist plot displays the electrochemical impedance of the samples, revealing that Te-Co3O4-α exhibits a smaller semicircle radius compared to Co3O4 (Fig. S9a in Supporting information), indicating that Te doping significantly enhances the specific conductance of the catalyst. The cyclic voltammetry (CV) curves (Fig. S9b in Supporting information) demonstrate that Te-Co3O4-α possesses higher redox peak currents, suggesting more efficient reduction of Co3+ to Co2+. Also, a smaller peak potential difference between the redox peaks suggests a lower kinetic barrier for the Co3+ → Co2+ transition. Above outcomes further confirm that Te-Co3O4-α can provide higher electron transfer efficiency for a faster Co3+/Co2+ valence state cycling ability during the activation reaction [40].
By analyzing the catalyst's surface chemical properties before and after the reaction, further details of the PMS activation process were explored. From Fig. 3d and Table S1, the proportion of Co3+/Co2+ increased from 0.53 to 0.68, indicating that Co2+ plays a key role in activating PMS. This corroborates the earlier conclusion that metal sites are the primary active centers. Meanwhile, after the activation reaction, the binding energy of Te 3d5/2 increased by 0.18 eV (Fig. 3e), suggesting a decrease in the electron cloud density around Te atoms and a conversion of Te species to a higher oxidation state [41]. This is likely because, as electron donors, the interstitial doped Te sites facilitate electron transfer to the Te-Co3O4-α surface during PMS activation to promote the reduction of Co3+ to Co2+, while the electrons around Te are consumed or redistributed. Additionally, the comparison of O 1s XPS spectra revealed a 14.7% decrease in OV content after the reaction, confirming the OV participation in PMS activation (Fig. 3f). The slight decrease in hydroxyl–oxygen content may be due to the participation of surface hydroxyl group for the capture of partial HSO5−, leading to their consumption. The lattice oxygen content significantly increases after the reaction, matching well with Zhao's report [42]. This indicates an increase in the stability of metal oxides, resulting in lower Co ions leaching of Te-Co3O4-α.
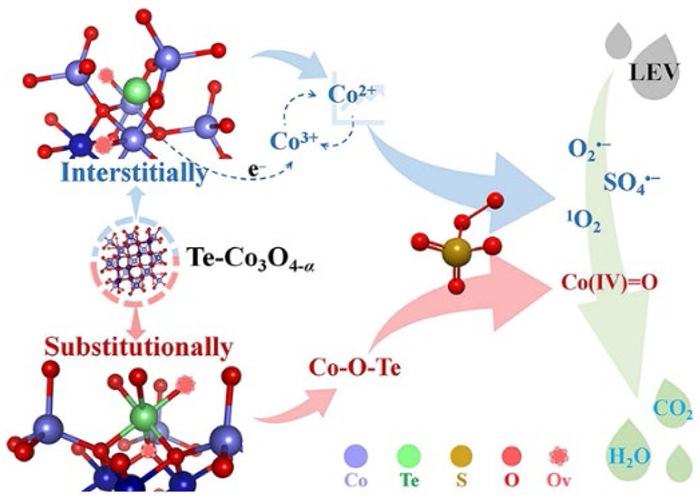
Based on the above discussions, the activation mechanism in the Te-Co3O4-α/PMS system could be reasonably speculated (Scheme 1). First, the interstitially doped Te induces lattice distortion of Co3O4 to form a large number of OV sites. This significantly increases the specific surface area and the density of low-valent Co species. As the primary active centers, the exposed Co sites react with the adsorbed PMS molecules, greatly enhancing the generation of radicals (Eqs. 6 and 7). In this process, Co3+ is reduced to Co2+ through redox reactions, completing the valence cycle. OV promotes the yield of active species by lowering the surface adsorption energy of PMS ions and the energy barrier for the O-O bond dissociation, while acting as an electron acceleration site and collaborating with interstitially doped Te4+ to overcome the rate-limiting step of the cycle (Eqs. 8 and 9). O2•− is likely generated from the unstable SO52− decomposition (Eq. 10) [31], while the production of 1O2 originates from the evolution of O2•− (Eqs. 11 and 12), or is from the dimerization reaction of SO5•− (Eqs. 8, 13 and 14) [33]. Additionally, the formation of HVMO is closely related to the electronic density around the metal atoms [37]. When substitution doping occurs, Te4+ replaces Co3+ to form a portion of the Co-O-Te site. Due to the higher electronegativity of Te4+, the electron cloud density near Co-O is reduced, thereby enhancing the electron delocalization at the Co sites [25]. This process weakens the filling of extra electrons in the Co 3d orbital. When the active center interacts with PMS, a metastable Co(Ⅱ)-OOSO3 complex is formed (Eq. 15). As the O-O bond breaks, a non-radical species, like Co(Ⅳ)=O, is easily formed through a two-electron transfer process (Eq. 16) [31].
|
|
(6) |
|
|
(7) |
|
|
(8) |
|
|
(9) |
|
|
(10) |
|
|
(11) |
|
|
(12) |
|
|
(13) |
|
|
(14) |
|
|
(15) |
|
|
(16) |
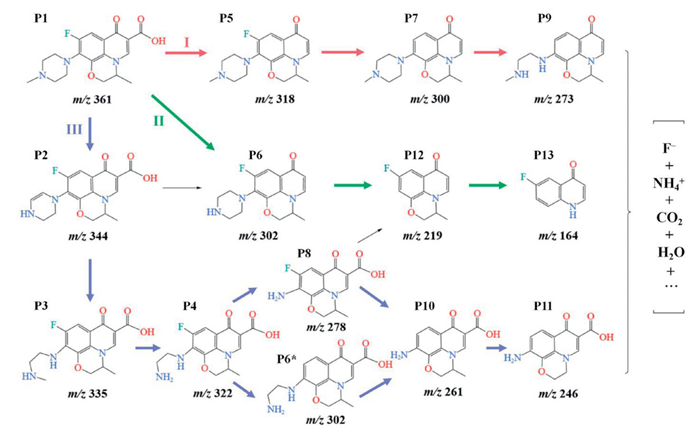
The Te-Co3O4-α/PMS system displayed high performance for removing LEV. The analysis of the treated supernatant by LC-MS provided insights into the degradation process and the intermediates (see analysis conditions in Table S3 in Supporting information). The molecular formulas of the intermediates obtained from the analysis, along with their corresponding secondary mass spectra, are presented in Fig. S10 and Table S4 (Supporting information), respectively. Based on these analytical results, Scheme 2 displays three possible primary degradation pathways. In pathway Ⅰ, the LEV molecule initially undergoes decarboxylation to form P5 (m/z 318). Subsequently, the C-F bond is selectively attacked by ROS, leading to the removal of a F− and the formation of P7 (m/z 300). Following this, P7 undergoes oxidative decomposition of the piperazine ring, yielding P9 (m/z 273). In pathway Ⅱ, P1 simultaneously loses both a methyl group and a carboxyl group to form P6 (m/z 302). After the piperazine ring undergoes hydroxylated and denitrogenated via ROS attack, the piperazine bond breaks to form P12 (m/z 219), which further decomposes through a ring-opening effect to produce P13 (m/z 164). In pathway Ⅲ, P1 first undergoes demethylation and oxidation of the piperazine ring to form P2 (m/z 344), P3 (m/z 335), and P4 (m/z 322). These intermediates are then further oxidized and defluorinated, yielding P8 (m/z 278), P6* (m/z 302) and P10 (m/z 261), followed by a demethylation to form P11 (m/z 246). Ultimately, the intermediate products are gradually decomposed into simple molecules, such as CO2 and H2O.
The effects of inorganic anions in water (i.e., Cl−, NO3−, SO42−, HCO3− and H2PO4−) on the LEV removal in the Te-Co3O4-α/PMS system at 1 and 5 mmol/L levels [33] were investigated. As shown in Fig. S11a (Supporting information), both lower and higher concentrations of Cl⁻ weaken the degradation process of LEV, reducing the removal to 74.0% and 73.0%, respectively. This is likely due to the reaction between Cl⁻ and PMS molecules, forming lower oxidative capability HClO (Eq. 17) [43]. Alternatively, it may be due to the reaction of Cl⁻ with the generated SO4•− (Eq. 18), which not only scavenges SO4•− but also produces radicals with weaker oxidative capacity (Eq. 19) [44]. The effects of NO3− and SO42− on LEV removal efficiency are minor (Figs. S11b and c in Supporting information). A slight inhibitory effect on the kinetics is likely attributed to the formation of NO3• with weak oxidizing ability (Eq. 20). The impact of SO42− is primarily due to its competitive adsorption with PMS on the surface hydroxyl groups of transition metal oxides [45]. At lower content, HCO3− has a negligible impact on LEV removal, while at higher HCO3− level, the LEV removal significantly decreases to 57.1% (Fig. S11d in Supporting information). The impact of HCO3− on LEV removal is likely via altering the acid-base environment of the solution. However, as shown in Table S5 (Supporting information), at the initial solution pH of 4.0 to 10.0, the final solution pH values were in the 3.18 to 3.88 range. This implies the minor effect of the initial solution pH on the LEV degradation in the Te-Co3O4-α/PMS system. Thus, the strong inhibition at higher HCO3− concentration is due to the excessive scavenging effect of HCO3− on radicals [6]. This not only generates radicals with weaker oxidizing ability but also depletes the original SO4•−, thereby limiting LEV removal (Eq. 21). Interestingly, we observed that the introduction of a low concentration of HCO3− markedly accelerated the reaction rate within the first ten minutes (Fig. S11d). This is due to the direct activation of PMS by HCO3−, which enhances the early-stage kinetic intensity [46]. The presence of H2PO4− had no significant effect on the removal of LEV, and its inhibitory effect on the interference kinetics was almost negligible (Fig. S11e in Supporting information). This is likely because the doping of Te changes the surface chemical properties of Co3O4 to form a stronger Co-O-Te bond that well limits the contact between H2PO4− and Co sites, thus preventing this chelating effect.
|
|
(17) |
|
|
(18) |
|
|
(19) |
|
|
(20) |
|
|
(21) |
The performance of the Te-Co3O4-α/PMS system for LEV removal in various actual water matrices is essential to evaluate its application potential. Natural organic matter (NOM) is widely present in aquatic environments and significantly influences pollutant degradation processes. As a complex natural polymer, humic acid (HA) contains multiple active functional groups (e.g., hydroxyl, carboxyl, ketone and amine) [47], its effect was investigated. As displayed in Fig. S12 (Supporting information), the removal of LEV decreased slightly by 8.70% in the presence of HA, indicating its minor influence in the Te-Co3O4-α/PMS system. Due to its abundant electron-donating groups, HA interacts with the electrophilic PMS through electron transfer reactions, consuming PMS, or occupies active surface sites via π-π interactions [48,49], thereby weakening LEV degradation. We further tested the performance of the Te-Co3O4-α/PMS system in different real waters. As shown in Fig. S12, the LEV degradation kept over 90% in tap, Jialing River, and Chongde Lake waters, highlighting the application potential of the Te-Co3O4-α/PMS system in real waters.
Generalizability testing provides a robust evaluation of the comprehensive degradation capability of advanced oxidation systems, offering insights into their applicability to various fluoroquinolone (FQ) targets. As traced in Fig. S13a (Supporting information), the removal rates of norfloxacin (NOR) and ciprofloxacin (CIP) exceed 80%, while those of gatifloxacin (GAT) and pefloxacin (PEF) achieve over 90%. The results indicate that the Te-Co3O4-α/PMS system exhibits good generalizability for FQs antibiotics. LEV mineralization capability in the Te-Co3O4-α/PMS system was acquired through measuring total organic carbon (TOC). TOC removal rate increases with PMS concentration. For instance, ~73% LEV can be mineralized at 3 mmol/L PMS level (Fig. S13b in Supporting information). Recyclability is another critical criterion for evaluating heterogeneous catalysts. As shown in Fig. S13c (Supporting information), when the used Te-Co3O4-α was reintroduced into the reaction system under identical conditions, above 90% LEV removal could be remained after the fifth cycle, demonstrating the catalyst's excellent stability and reusability. Finally, we compared the normalized degradation kinetic constants of other non-phase conversion doping-based Co3O4 catalysts (circle) [8–10,16,26,31,42,50–54] and heterogeneous catalytic degradation systems targeting LEV (rhombus) [55–68]. As traced in Fig. S13d (Supporting information), under identical mass loading of catalyst, the Te-Co3O4-α/PMS system achieves significantly higher degradation kinetics even at a lower PMS dosage (star), surpassing the performance of previously reported systems. This demonstrates that the Te-Co3O4-α/PMS system in this study exhibits outstanding degradation performance, highlighting its strong potential for practical applications.
In summary, via a simple one-step hydrothermal method, Te was doped into Co3O4 in two modalities, i.e., interstitial and substitutional. The acquired Te-Co3O4-α exhibited highly efficient PMS catalytic activity. In the Te-Co3O4-α/PMS system, the removal rate of LEV reached 96.5%, with a degradation kinetic constant of 0.2129 min−1, which is 66 times higher than that of the Co3O4/PMS system. The coexistence of doping modalities induces lattice expansion and local electron rearrangement, modulating the specific surface area and metal active centers. The formation of SO4•−, O2•−, and 1O2 primarily originates from the effect of interstitial Te sites on Co active centers, while that of high-valent Co(Ⅳ)=O is mainly driven by the interaction between substitutional Co-O-Te sites and PMS. The contribution of •OH is minor, and the ETP does not occur in the system. In addition, the Te-Co3O4-α/PMS system exhibits wide pH adaptability, generalizability for FQs antibiotics, and good tolerance to various water matrices, indicating its good practical application potential. This study enriches the variety of doped-metal oxide catalysts for PMS activation, elucidating the synergistic modulation of the electronic structure triggered by the coexistence of a dual-doping configuration. It also highlights the correlation between coexisting doping modalities and different ROS generation pathways, and clarifies the possibly overestimated "production role" of OV in metal oxides. These findings hold significant importance for developing transition metal spinel oxide catalysts based on non-phase-transition doping and for the application of oxygen vacancy engineering-driven advanced oxidation systems.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Jianke Tan: Writing – original draft, Methodology, Investigation, Data curation. Xiaodan Zhang: Writing – review & editing, Supervision, Project administration. Yuming Huang: Writing – review & editing, Supervision, Methodology, Funding acquisition, Conceptualization.
This work is supported by the National Natural Science Foundation of China (No. 42177054).
Supplementary material associated with this article can be found, in the online version, at doi:
X. Pan, J. Ji, N. Zhang, M. Xing, Chin. Chem. Lett. 31 (2020) 1462–1473. doi: 10.1016/j.cclet.2019.10.002
X. Liu, S. Wang, X. Wang, Chin. Chem. Lett. 36 (2025) 110679. doi: 10.1016/j.cclet.2024.110679
C. Zuo, X. Tai, Z. Jiang, et al., Molecules 28 (2023) 3495. doi: 10.3390/molecules28083495
N. Li, J. Ye, H. Dai, et al., Water Res. 235 (2023) 119926. doi: 10.1016/j.watres.2023.119926
X. Li, C. Huang, W. Han, T. Ouyang, Z. Liu, Chin. Chem. Lett. 32 (2021) 2597–2616. doi: 10.1016/j.cclet.2021.01.047
P. Hu, M. Long, Appl. Catal. B 181 (2016) 103–117. doi: 10.1016/j.apcatb.2015.07.024
Q. Tian, B. Yu, Z. Li, et al., Chin. Chem. Lett. 36 (2025) 110322. doi: 10.1016/j.cclet.2024.110322
M. Lu, G. Kang, Y. Deng, CrystEngComm 25 (2023) 2767–2777. doi: 10.1039/d3ce00067b
H. Zhang, Q. An, Y. Su, X. Quan, S. Chen, J. Hazard. Mater. 448 (2023) 130987. doi: 10.1016/j.jhazmat.2023.130987
X. Zhang, S. Chen, W. Zhang, X. Ma, Sep. Purif. Technol. 353 (2025) 128648. doi: 10.1016/j.seppur.2024.128648
Z. Liu, Y. Zhong, L. Chen, et al., J. Hazard. Mater. 478 (2024) 135437. doi: 10.1016/j.jhazmat.2024.135437
F. Li, C. Yuan, Y. Niu, et al., Environ. Res. 263 (2024) 120249. doi: 10.1016/j.envres.2024.120249
L. Li, L. Song, X. Zhang, S. Zhu, Y. Wang, ACS Appl. Energy Mater. 5 (2022) 2505–2513. doi: 10.1021/acsaem.1c04033
M. Fan, C. Wang, X. Yu, et al., Chem. Eng. J. 487 (2024) 150751. doi: 10.1016/j.cej.2024.150751
Y. Su, W. Sun, L. Yuan, et al., J. Water Process Eng. 64 (2024) 105574. doi: 10.1016/j.jwpe.2024.105574
X. Zhou, Z. Tang, M. Du, et al., Sep. Purif. Technol. 354 (2025) 129434. doi: 10.1016/j.seppur.2024.129434
T. Zhang, S. Wu, N. Li, G. Chen, L. Hou, J. Hazard. Mater. 449 (2023) 130971. doi: 10.1016/j.jhazmat.2023.130971
Y. Bu, H. Li, W. Yu, et al., Environ. Sci. Technol. 55 (2021) 2110–2120. doi: 10.1021/acs.est.0c07274
Q. Qin, T. Liu, J. Zhang, et al., J. Hazard. Mater. 419 (2021) 126447. doi: 10.1016/j.jhazmat.2021.126447
S. Liu, H. Fu, F. Wang, et al., Appl. Catal. B 346 (2024) 123753. doi: 10.1016/j.apcatb.2024.123753
L. Kong, G. Fang, Z. Fang, et al., Chem. Eng. J. 416 (2021) 128996. doi: 10.1016/j.cej.2021.128996
Y. Hao, B. Liu, L. Tian, et al., ACS Appl. Mater. Interfaces 9 (2017) 12687–12693. doi: 10.1021/acsami.6b16856
A. Torma, W. Li, H. Zhang, et al., ACS Nano 15 (2021) 20550–20561. doi: 10.1021/acsnano.1c09142
J. Yang, M. Zheng, Y. Wu, et al., Appl. Catal. B 343 (2024) 123478. doi: 10.1016/j.apcatb.2023.123478
M. Xu, T. Zhao, X. Yang, et al., Sep. Purif. Technol. 359 (2025) 130503. doi: 10.1016/j.seppur.2024.130503
Q. Gao, H. Li, X. Wang, et al., Appl. Surf. Sci. 574 (2022) 151632. doi: 10.1016/j.apsusc.2021.151632
E. Annese, F. Stavale, Appl. Surf. Sci. 672 (2024) 160710. doi: 10.1016/j.apsusc.2024.160710
Y. Wan, Z. Li, X. Zheng, et al., J. Colloid Interface Sci. 663 (2024) 177–190. doi: 10.1016/j.jcis.2024.02.149
M. Ding, W. Ao, H. Xu, et al., J. Hazard. Mater. 420 (2021) 126686. doi: 10.1016/j.jhazmat.2021.126686
Y. Huang, Y. Huang, C. Huang, C. Chen, J. Hazard. Mater. 170 (2009) 1110–1118. doi: 10.1016/j.jhazmat.2009.05.091
Y. Chen, D. Chen, X. Bai, Chem. Eng. J. 479 (2024) 147886. doi: 10.1016/j.cej.2023.147886
J. Liang, L. Fu, K. Gao, Appl. Catal. B 315 (2022) 121542. doi: 10.1016/j.apcatb.2022.121542
J. Tan, L. Chang, X. Zhang, H. Chai, Y. Huang, Sep. Purif. Technol. 357 (2025) 130034. doi: 10.1016/j.seppur.2024.130034
Z. Guo, R. Sun, Z. Huang, W. Li, Proc. Natl. Acad. Sci. 120 (2023) e2220608120. doi: 10.1073/pnas.2220608120
Y. Li, M. Fan, C. Wang, et al., Chin. Chem. Lett. 35 (2024) 109764. doi: 10.1016/j.cclet.2024.109764
S. Zhu, H. Li, L. Wang, et al., Chem. Eng. J. 458 (2023) 141415. doi: 10.1016/j.cej.2023.141415
S. Liu, J. Du, H. Wang, et al., Water Res. 254 (2024) 121417. doi: 10.1016/j.watres.2024.121417
J. Yu, Z. Gong, S. Wang, et al., Appl. Catal. B 364 (2025) 124850. doi: 10.1016/j.apcatb.2024.124850
L. Kong, G. Fang, Y. Chen, et al., Appl. Catal. B 253 (2019) 278–285. doi: 10.1016/j.apcatb.2019.04.069
S. Zuo, Y. Ding, L. Wu, et al., Water Res. 231 (2023) 119631. doi: 10.1016/j.watres.2023.119631
X. Li, R. Lv, W. Zhang, et al., Water Res. 228 (2023) 119363. doi: 10.1016/j.watres.2022.119363
Z. Zhao, L. Zhang, F. Qian, et al., Langmuir 41 (2025) 1024–1036. doi: 10.1021/acs.langmuir.4c04333
J. Yan, L. Gong, S. Chai, et al., Sep. Purif. Technol. 302 (2022) 122016. doi: 10.1016/j.seppur.2022.122016
Y. Zhao, X. Yuan, L. Jiang, et al., Chem. Eng. J. 400 (2020) 125903. doi: 10.1016/j.cej.2020.125903
Y. Han, C. Zhao, W. Zhang, et al., Appl. Catal. B 340 (2024) 123224. doi: 10.1016/j.apcatb.2023.123224
L. Jaesang, G. Urs, K. Jae, Environ. Sci. Technol. 54 (2020) 3064–3081. doi: 10.1021/acs.est.9b07082
W. Chen, Z. Liu, Y. Xie, et al., Int. J. Biol. Macromol. 298 (2025) 139700. doi: 10.1016/j.ijbiomac.2025.139700
S. Li, C. Dai, L. Wan, et al., Sep. Purif. Technol. 361 (2025) 131564. doi: 10.1016/j.seppur.2025.131564
J. Li, J. Wei, J. Sun, et al., Chem. Eng. J. 505 (2025) 159739. doi: 10.1016/j.cej.2025.159739
W. Zhang, L. Chang, J. Tan, et al., Sep. Purif. Technol. 327 (2023) 124933. doi: 10.1016/j.seppur.2023.124933
H. Li, Q. Gao, G. Wang, et al., Chem. Eng. J. 426 (2021) 131292. doi: 10.1016/j.cej.2021.131292
X. Zhang, S. Liu, Z. Wang, et al., Chem. Eng. J. 477 (2023) 146987. doi: 10.1016/j.cej.2023.146987
X. Wang, X. Luo, R. Li, et al., Chem. Eng. J. 491 (2024) 152197. doi: 10.1016/j.cej.2024.152197
X. Song, Y. Zhou, W. He, et al., J. Mater. Sci. Mater. Electron. 35 (2024) 851. doi: 10.1007/s10854-024-12491-x
F. Ul Rehman, A. Iqbal, A. Khalid, et al., J. Mol. Liq. 404 (2024) 124978.
J. Gu, L. Zhang, Y. Ji, et al., Mater. Res. Bull. 182 (2025) 113143.
Q. Zhao, Y. Wu, X. Zhang, et al., Appl. Surf. Sci. 657 (2024) 159716.
W. Liu, S. Zhou, W. Xu, et al., J. Water Process Eng. 70 (2025) 107101.
X. Song, H. Zhang, X. Diao, et al., Dyes Pigments 232 (2025) 112467.
M. Chen, S. Zhuo, W. Zhao, et al., Appl. Surf. Sci. 679 (2025) 161161.
C. Zhao, L. Xu, Y. Wang, et al., Chem. Eng. J. 496 (2024) 153712.
W. Zhang, B. He, J. Shang, et al., Chem. Eng. J. 506 (2025) 160316.
M. Cheng, X. Long, P. Song, et al., Chem. Eng. J. 480 (2024) 147885.
C. Wang, L. Chang, X. Zhang, H. Chai, Y. Huang, Environ. Res. 252 (2024) 118892.
Q. Zhao, D. Liu, Y. Wu, et al., Sep. Purif. Technol. 352 (2025) 128089.
Z. Liu, J. Wu, K. Li, C. Song, X. Guo, Ind. Eng. Chem. Res. 63 (2024) 9082–9092. doi: 10.1021/acs.iecr.4c00877
X. Yu, S. Wang, Y. Zhang, et al., Small 21 (2025) 2410819.
C. Ruan, L. Zhang, Z. Song, et al., ChemistrySelect 10 (2025) e202405300.

Figure 1 (a) XRD patterns of prepared samples, (b) locally amplified XRD patterns, (c) total XPS spectra, (d) SEM image, (e) TEM image, (f) HR-TEM image and (g-k) elements mapping of Te-Co3O4-α.

Figure 2 (a) Adsorption/degradation kinetics of LEV in each system. Trial conditions: 10 mg/L LEV, 1.0 mmol/L PMS, 20 mg/L of catalysts, initial pH 6.6 ± 0.1, 25 ℃. Effects of (b) Te doping amount, (c) catalysts dosage, (d) PMS concentration, (e) initial solution pH, and (f) reaction temperature on LEV degradation. Insets are corresponding apparent rate constants.

Figure 3 EPR spectra of (a) DMPO-SO4•− and DMPO-•OH, (b) DMPO-O2•−, and (c) 2,2,6,6-tetramethylpiperidine (TEMP)-1O2 in the Te-Co3O4-α/PMS system. Trial conditions: 100 mmol/L DMPO and TEMP, 1.0 mmol/L PMS, 20 mg/L of catalysts, initial pH 6.6 ± 0.1, 25 ℃. High-resolution XPS spectra comparison of (d) Co 2p, (e) Te 3d, and (f) O 1s of the fresh and used Te-Co3O4-α.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:


 下载:
下载: