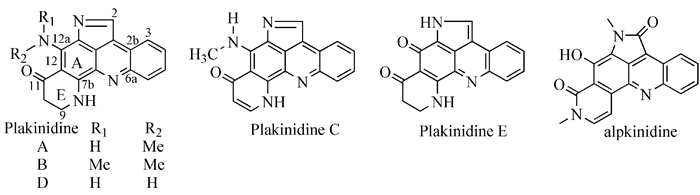
 图式1
普莱克尼啶类化合物的结构图
图式1.
Structures of Plakinidines
图式1
普莱克尼啶类化合物的结构图
图式1.
Structures of Plakinidines
引用本文:
陈思宏, 刘殿卿, 谭芸妃, 郑小刚. 普莱克尼啶及其类似生物碱的研究进展[J]. 化学通报,
2017, 80(8): 725-733.

Citation: Chen Sihong, Liu Dianqing, Tan Yunfei, Zheng Xiaogang. Research Progresses in Plankinidines and Analogous Alkaloids[J]. Chemistry, 2017, 80(8): 725-733.
Citation: Chen Sihong, Liu Dianqing, Tan Yunfei, Zheng Xiaogang. Research Progresses in Plankinidines and Analogous Alkaloids[J]. Chemistry, 2017, 80(8): 725-733.
普莱克尼啶及其类似生物碱的研究进展
English
Research Progresses in Plankinidines and Analogous Alkaloids
Abstract:
Plakinidines and analogues are the pyrrolacridine and pyridoacridine polycyclic alkaloids that are isolated from ocean mollusks. And plakinidines are the first class of pyrrolacridine containing alkaloids found in marine extracts, some of which have good biological activities such as anti-cancer and anti-parasite. In this paper, studies on the extraction, isolation, structural elucidation, bio-activity and chemical synthesis of plakinidines and analogous alkaloids are reviewed.
-
Key words:
- Plakinidines
- / Alkaloids
- / Biological activity
- / Chemical synthesis
-
1969年Weinheimer等[1]从佛罗里达海域柳珊瑚(Plexaura homomalla)中分离出前列腺素15R-PGA2,成为“海洋药物”开发的里程碑。人类为了寻找新型抗癌、抗艾滋病和抗心脑血管疾病药物,对海洋生物的研究越来越广泛。迄今为止,在海洋生物中发现了多种活性物质,如前列腺素(Prostaglandins)[2~4]、膜海鞘素(Didemnin B)[5]、片螺素(Lamellarin)[6, 7]等。1990年,Inman等[8]从Plakortis海绵中提取出两种结构新颖的多芳香吡咯并吖啶稠环生物碱,命名为Plakinidine A和B(本课题组将Plakinidine暂译为普莱克尼啶)。近年来,我国对海洋生物的研究日渐活跃,但尚未见国内对此类海洋活性物质的提取和全合成报道。
目前已发现的普莱克尼啶及其类似化合物主要有两种结构:一种是吡咯并吖啶环中心结构,这种体系化合物的报道极少,它们分别是Plakinidine A~E[8~12]和Alpkinidines[13, 14] (图式 1);另一种是吡啶并吖啶环中心结构,如amphimidine、pantherinine、cystodytins、styelsa-mines、neoamphimidine、deoxyamphimidine, arnoa-mines A和B、sherliamine D和E、cyclosherliamine、eilatin、stelletamine (图式 2)。
图式1
普莱克尼啶类化合物的结构图
图式1.
Structures of Plakinidines
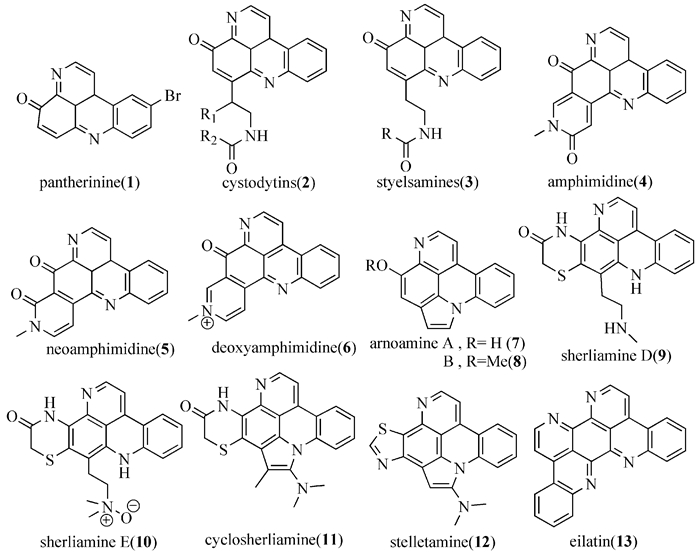 图式2
普莱克尼啶类似生物碱的结构
图式2.
Sructures of analogous alkaloids of Plakinidines
图式2
普莱克尼啶类似生物碱的结构
图式2.
Sructures of analogous alkaloids of Plakinidines
1 普莱克尼啶及其类似生物碱的提取、分离和结构确定
1.1 普莱克尼啶生物碱的提取和分离
1990年,Inman等[8]在一种名为Plakortis海绵的海洋生物中用甲醇提取到红色粘稠油状物,分别用环己烷、四氯化碳、二氯甲烷萃取,用柱色谱进一步分别分离四氯化碳、二氯甲烷萃取液得两种化合物,一种是紫色的固体普莱克尼啶A,另一种是紫色的油状物普莱克尼啶B。与此同时,犹他州大学的West等[9]不仅分离出普莱克尼啶A和B,还得到了普莱克尼啶C。1997年,Smith等[10]用乙醇提取red Didemnum海绵,提取液用氯仿萃取,经柱色谱、凝胶色谱分离得红色固体普莱克尼啶D,同一时期,Ford等[11]也发现了普莱克尼啶D。2007年,Ralifo等[12]采用溶剂萃取Plakortis quasiamphiaster,萃取液经柱色谱分离出24份样品,其中第19份进一步分离得到了暗红色固体普莱克尼啶E(图 1)。
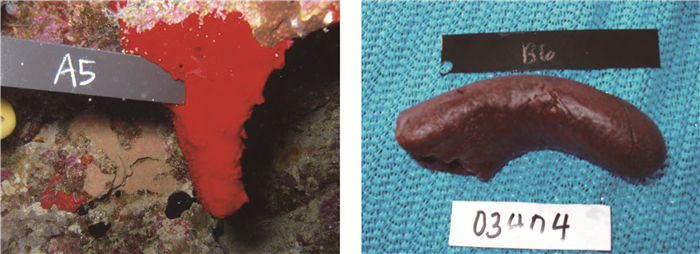 图 1
Plakortis quasiamphiaster采集实物图
Figure 1.
Underwater (left) and abovewater (right) pictures of Plakortis quasiamphiasterb
图 1
Plakortis quasiamphiaster采集实物图
Figure 1.
Underwater (left) and abovewater (right) pictures of Plakortis quasiamphiasterb
1.2 普莱克尼啶类似生物碱的提取和分离
1993年,Kim等[13]用甲醇对澳大利亚豪斯湾的Aplidium pantherinum进行提取,进一步用氯仿/甲醇提取甲醇中的提取物,得到Pantherinine(1) 粗品,再用半制备色谱即可将其纯化,洗脱剂为甲醇:水=7:3(体积比),该物质分子式为C15H8N3OBr。1983年,Schmitz等[15]室温下分别用二氯甲烷、氯仿和甲醇、甲醇提取一种Amphi-medon海绵,再用氯仿热索式提取样本,最后经氧化铝柱色谱(氯仿和甲醇为洗脱剂)得到黄色固体Amphimidine (4),其分子式为C19H11N3O2。1998年,Plubrukarn等[16]首次报道Arnoamines A和Arnoamines B的分离与结构确定,他们用甲醇提取经均匀浸泡、冷冻干燥的一种Cystodytes海绵,得到的样品分别经正反相柱色谱提纯即可得到纯的Arnoamines A和Arnoamines B,并确定它们的分子式分别为C17H10N2O和C18H12N2O。1998年,Koren-Goldshlager等[17, 18]从马达加斯加西北的印度洋海鞘中分离出含硫杂环结构sherliamine D和E、cyclosherliamine三种生物碱。Rudi等[19]也曾经报道了一种结构更复杂的类似生物碱eilatin。
1.3 普莱克尼啶及其类似生物碱的结构确定
HREIMS和APT 13C NMR测得普莱克尼啶A分子式为C18H14N4O (m/z 302.1169,[M+])[8],确定普莱克尼啶是含有吡咯并吖啶结构的生物碱。普莱克尼啶A红外光谱在1624cm-1处有吸收峰,13C谱显示C-11在δ194 (DMSO-d6)处有信号,断定普莱克尼啶A分子结构中含有一个羰基。普莱克尼啶B分子式确定为C19H16N4O,MS (m/z 316.1322,[M+])[8],1H和13C NMR化学位移和自旋耦合与普莱克尼啶A非常相似,低温核磁共振谱显示N原子上有两个甲基[13C (δ 47.3,45.0),1H (δ 3.92,3H;3.32,3 H)],即普莱克尼啶B是A的N-甲基化物。普莱克尼啶C分子式是C18H12N4O,1H和13C NMR δC9、δC10比普莱克尼啶A相应的C9、C10的化学位移大很多,表明普莱克尼啶C在C9、C10之间形成双键,是普莱克尼啶A的9、10位去氢化物。普莱克尼啶D分子式是C17H12N4O (m/z 288.1048)[11],其核磁共振数据与普莱克尼啶A、B、C非常相似,且一些物理性质(如颜色等)也很相似,它是普莱克尼啶A的N-13去甲基化合物。普莱克尼啶E经HRMS探测到其分子式为C17H11N3O2(m/z 290.0933),核磁试验数据显示普莱克尼啶E比D少了一个-NH2而多了一个-O-。
普莱克尼啶及其类似生物碱分别含有如图式 3所示的吡咯并吖啶、吡啶并吖啶环骨架结构,对于这类化合物的结构确定,可用共性的质谱、核磁特征作为指导。通常用EI源高分辨率质谱对其进行分析,其分子离子峰M+是该类化合物失去一个电子形成的,一般出现在质谱图的最右侧且丰度比较低;其碎片离子峰也比较少,主要是连在该骨架结构上的链或环断裂形成碎片离子。这类化合物的1H NMR有以下共同特点:在δ 7~9附近会出现4个多重峰,这对应着它们骨架结构C环上的4个氢。对于普莱克尼啶类化合物,在δ 8~9之间还会出现一个单峰,这对应着D环中的1个氢;在δ10附近出现一个宽的单峰和δ 2~4之间出现2个三重峰,这些对应着E环上的所有氢。而对于普莱克尼啶类似生物碱,在δ 8~9附近还会出现2个双重峰,这对应着D环上的2个氢。至于这类物质的13C NMR谱,如果δ大于160处有峰,则该生物碱可能含有羰基;在δ 130附近出现4个叔碳峰,这对应着C环上的4个叔碳;在δ 115~150之间出现的5个季碳峰,则对应着B环上的5个季碳。普莱克尼啶类化合物在δ 120~140之间出现的一个叔碳峰,对应着D环上的一个叔碳。而普莱克尼啶类似生物碱在δ 110~140之间出现D环上的两个叔碳峰。这类生物碱谱图的共性可用于鉴定该类化合物的结构,但准确的结构鉴定还需要二维核磁波谱、红外等多种手段,有的甚至需要X射线衍射来确定(如alpinidine、eliatin(13))。
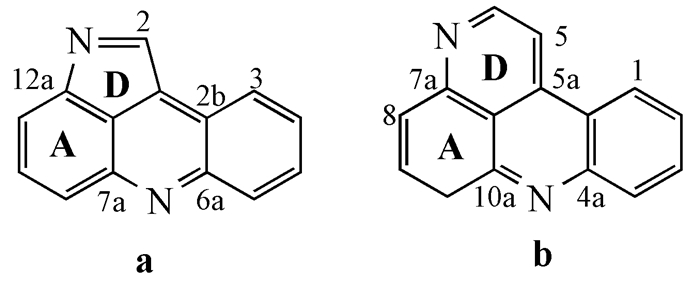 图式3
普莱克尼啶及其类似物的骨架
图式3.
The skeleton of Plakinidines and analogues
图式3
普莱克尼啶及其类似物的骨架
图式3.
The skeleton of Plakinidines and analogues
2 普莱克尼啶生物碱的生物活性研究
含有普莱克尼啶的海洋生物非常罕见,关于普莱克尼啶生物活性的研究报道尚不多见。但在普莱克尼啶结构确定的同时,科学家对普莱克尼啶进行的初步活性研究证明普莱克尼啶具有较好的抗癌和杀菌能力。
West等[9]研究表明,普莱克尼啶A、B、C针对鼠类白血病的IC50分别为0.1、0.3、0.7 μg/mL。Inman等[8, 20]报道普莱克尼啶A、B在体外具有抗巴西钩虫的功能、普莱克尼啶A对逆转录酶有抑制作用[8, 21]。Smith等[10]报道普莱克尼啶D在5μg/mL浓度下具有体外抗人体结肠癌细胞作用。
2007年,Ralifo等[12]发现普莱克尼啶A对人体H-116细胞具有选择性抑制作用,IC50为0.23μg/mL。克隆原细胞存活寿命实验结果显示普莱克尼啶A连续作用2、24和168 h,使存活率降到10%的普莱克尼啶A的相应浓度为20μg/mL、1.5μg/mL、150 ng/mL(图 2)。这意味着,癌症患者每天摄入极少量普莱克尼啶A就有很好的治疗作用。酵母菌琼脂圆环试验显示普莱克尼啶E有抑制啤酒酵母菌拓扑酶I的毒性作用,IC50为3.30×10-8 mol/L,而对野生型酵母菌没有毒性。
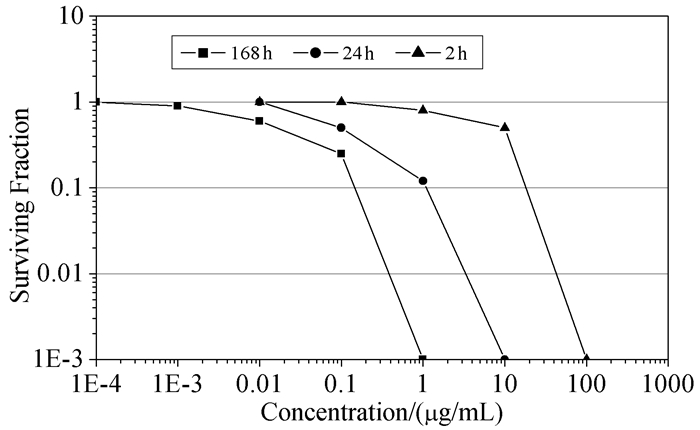 图 2
普莱克尼啶A抑制H-116细胞作用示意图
Figure 2.
Cytotoxicity of Plakinidine A against H-116 cells
图 2
普莱克尼啶A抑制H-116细胞作用示意图
Figure 2.
Cytotoxicity of Plakinidine A against H-116 cells
3 普莱克尼啶及其类似生物碱的化学合成研究
海洋天然活性物质成分含量低、活性强,直接以海洋生物为原料进行活性物质提取分离很难满足需求,加之大量捕获海洋生物会给海洋生态和环境带来严重的不良影响,因此,海洋活性物质的全合成工作具有重要意义[22]。作为人类发现的第一类吡咯并吖啶结构生物碱,普莱克尼啶不仅具有新颖的结构,而且表现出较好的生物活性,所以其全合成研究意义重大。目前开展普莱克尼啶全合成的研究较少,其中英国曼彻斯特大学和日本明治药科大学的研究较为活跃[23],但尚未见普莱克尼啶全合成的报道。为了深入研究普莱克尼啶类生物碱,探索其潜在的药学价值,笔者课题组近年来开展了普莱克尼啶的全合成研究,设计了一条较文献[23]简单易行的合成方案,并取得了一定进展[39, 40]。
3.1 普莱克尼啶及其中间体的合成研究
剖析普莱克尼啶的分子结构,可以得到4种主要的合成子:(1)5, 6-二氨基-1, 10-二氮杂菲;(2) 苯并[b]-1, 10-二氮杂菲及其衍生物;(3)3-氢吡咯并[3, 4-c]喹啉衍生物;(4) 吡咯并[2, 3-g]喹啉衍生物,如图式 4所示。这4种中间体都可以用来合成普莱克尼啶,前三种已有相关的合成文献,唯独未见第四个结构单元相关的合成文献,但有关于此结构合成的前体-4-硝基-2, 3-吲哚二酮的合成报道。
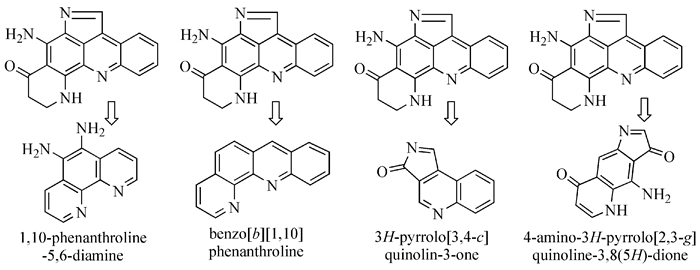 图式4
普莱克尼啶合成子
图式4.
The synthons of pyridoacridines
图式4
普莱克尼啶合成子
图式4.
The synthons of pyridoacridines
3.1.1 5, 6-二氨基-1, 10-二氮杂菲的合成研究
5, 6-二氨基-1, 10-二氮杂菲的合成方法有两种。一种是先将1, 10-二氮杂菲氧化为1, 10-二氮杂菲-5, 6-二酮,然后将其转化为肟,最后还原得到5, 6-二氨基-1, 10-二氮杂菲[24](式(1))。
另一种方法是将5-硝基-1, 10-二氮杂菲转化为5-氨基-6-硝基-1, 10-二氮杂菲,后者进一步还原得5, 6-二氨基-1, 10-二氮杂菲[25](式(2))。
方法一较方法二路线短,经三步反应以67%的收率得到目标产品,反应条件温和,操作简单;方法二反应路线长,反应条件苛刻,目标产物收率低。因此,方法一是目前合成5, 6-二氨基-1, 10-二氮杂菲比较好的选择。
3.1.2 苯并[b]-1, 10-二氮杂菲及其衍生物的合成研究
苯并[b]-1, 10-二氮杂菲及其衍生物8, 9, 10, 11-四氢苯并[b]-1, 10-二氮杂菲与普莱克尼啶的结构非常相似。目前对它们的合成有如下三种方法。
(1) 由4-氨基吖啶-3-甲醛和3-乙酰基-2, 4-戊二酮合成苯并[b]-1, 10-二氮杂菲[26](式(3))。该法反应条件温和,易于操作,但目标产物的选择性较低。
(2) 由邻溴苯甲酸与2-氨基喹啉合成12-氢苯并[b]-1, 10-二氮杂菲-7-酮[27] (式(4))。经两步反应产品收率达62%,但这两步反应都需要150℃的高温,且后处理麻烦,需酸碱中和以及柱色谱进行纯化。
(3) 由2-氨基-3-喹啉甲醛与环己酮合成8, 9, 10, 11-四氢苯并[b]-1, 10-二氮杂菲[28~31] (式(5))。该法合成路线短,产品收率高达86%,但反应条件比较苛刻,需使用强碱。
苯并[b]-1, 10-二氮杂菲类衍生物与普莱克尼啶结构非常相似,这类化合物的合成方案对于人工合成普莱克尼啶有重要意义。如利用上述合成5, 6-二氨基-1, 10-二氮杂菲的方法或其他方法,将其转化为5, 6-二氨基苯并[b]-1, 10-二氮杂菲,可以方便地合成出普莱克尼啶骨架(式(6))。
3.1.3 3-氢吡咯并[3, 4-c]喹啉衍生物的合成研究
3-氢吡咯并[3, 4-c]喹啉衍生物(式(7))通常以2, 3吲哚二酮为原料合成[32, 33]。此法原料价廉易得,最终目标产物收率能达43%~60%;其主要缺点是反应路线较长、操作繁琐、反应条件苛刻。
也有研究者[34]由2-氨基-3-氰基-4-喹啉甲酸直接合成(式(8))。该法反应条件温和,后处理简单,产品收率高达90%以上,但原料不易得。
3.1.4 4-硝基-2, 3-吲哚二酮的合成研究
1989年Parrick等[35]从4-硝基吲哚出发,经过三步反应合成了4-硝基-2, 3-吲哚二酮(式(9))。该路线反应条件温和、后处理比较简单,但反应路线较长,且目标产品收率较低(26%)。
将硝基还原为氨基,再与环己酮反应则可制得7, 8, 9, 10-四氢吡咯并[2, 3, 4-kl]吖啶(式(10)),这一化合物可以进一步合成普莱克尼啶类化合物,是合成普莱克尼啶的重要中间体之一。
3.1.5 6, 11-二氢苯并[b]吡咯并[4, 3, 2-de]-1, 10-二氮杂菲-5, 8-二酮合成研究
2004年,Kitahara等[23]首次报道了一种合成普莱克尼啶中间体—6, 11-二氢苯并[b]吡咯并[4, 3, 2-de]-1, 10-二氮杂菲-5, 8-二酮的方法(式(11))。2-乙酰基-3′-硝基二苯胺14在硫酸-醋酸中环化得到9-甲基-1-硝基吖啶15和9-甲基-3-硝基吖啶16(同分异构体分离非常困难(15与16的比例约为2:1))。直接还原得到9-甲基-1-氨基吖啶17和9-甲基-3-氨基吖啶18,从化合物14出发合成到17和18,产率分别是39%和11%。化合物17乙酰化得到19,再经二氧化锡氧化得环状化合物20。在0~5 ℃用硝酸钾和硫酸对20进行硝化得21,再经10%H2SO4脱去乙酰基生成22。21用亚硫酸钠还原得到化合物23,再与2, 2-二甲基-1, 3-二氧杂-4, 6-环己二酮和原甲酸三甲酯反应生成麦尔酮酸衍生物24,环化得中间体25,用20%硫酸脱去乙酰基得到化合物26。从24到26的产率为81%。此路线不足之处在于26硝化后,硝基易取代a位而不是b位,不能在指定位置引入含N基团,这对合成普莱克尼啶C极为不利,导致全合成未能成功。
3.2 普莱克尼啶类似生物碱的合成研究
近年来报道了多种与普莱克尼啶结构类似生物碱的全合成,这些化合物的全合成方案将为普莱克尼啶的合成工作提供指导。
1997年,Kitahara等[36]合成的Cystodamine的异构体,与普莱克尼啶的结构非常相似,其合成路线见式(12)。该法反应条件温和,但反应路线长,而且反应速率慢,目标产物收率相对较低(13%)。
2001年Delfourne等[37]合成了一系列吡啶并吖啶衍生物(式(13))。所采用的两条合成路线反应条件都较温和,操作简单,其主要缺点是反应速率慢、后处理过程繁琐且收率低。
2000年,Âlvarez等[38]合成了4-氨基-6氢-吡啶并吖啶-6酮(式(14))。经该化合物可进一步合成含吡啶并吖啶环类生物碱。该方法路线长、收率低,涉及低温反应,操作繁琐,后处理麻烦。
3.3 本课题组的普莱克尼啶化学合成研究
由于普莱克尼啶化合物具有新颖的化学结构,且表现出较好的生物活性,在多年从事海洋活性物质全合成工作的基础上,本组开展了普莱克尼啶的全合成研究工作,设计了简单易行的合成方案,取得了很好的进展,并合成出普莱克尼啶的几种重要中间体。
3.3.1 9-甲基-2, 4-二硝基-5-氯吖啶环的合成[39]
以间二氯苯为原料,硝化得1, 5-二氯-2, 4-二硝基苯,预先将N原子引入相应的位置,再经乌尔曼偶联、傅克关环、重排得普莱克尼啶的重要中间体9-甲基-2, 4-二硝基-5-氯吖啶(式(15))。
3.3.2 9-甲基-2, 4-二氨基-1-氯吖啶合成[40]
方法一:间二氯苯经过硝化,得到化合物27,与邻氨基苯乙酮反应得到化合物28,在三氟甲磺酸催化下关环得到29,在DBU(1, 8-二氮杂二环十一碳-7-烯)中重排得到30,还原得到31。
方法二:化合物28还原得到32,将32的氨基保护得到33,在浓硫酸作用下关环得到34,然后脱掉氨基保护基得到31(式(16))。
本课题组对这两种普莱克尼啶中间体的合成研究发现,影响这两种普莱克尼啶中间体收率的关键是化合物28的关环反应,化合物28很难发生关环反应得到化合物29。我们探索了在浓硫酸、三氯氧磷、三氟甲磺酸作用下化合物28的关环情况,发现使用浓硫酸和三氯氧磷时,化合物28均未发生关环;在三氟甲磺酸作用下,化合物28也只有少量的关环。这一关环反应关系到普莱克尼啶中间体的合成收率的高低,仍需化学工作者进一步深入研究。
4 结语
普莱克尼啶类化合物是一种结构新颖的化合物,这无疑又是人工合成化学结构的一项新的挑战。Tatsuta等[41]提到可以通过合成吡咯喹啉的方法来合成普莱克尼啶类化合物。本课题组从事普莱克尼啶的合成研究几年来,设计了几套简单易行的合成方案,并取得了一些重要突破。目前,这类化合物的全合成尚未实现,但基于该类化合物具有抗癌、杀菌等应用价值,因此对其进行全合成是一项意义重大且很具挑战性的工作,相信在广大相关科研工作者的共同努力下,普莱克尼啶的全合成在不久的将来就会完成。
-
-
[1]
A J Weinheimer, R L Spraggins. Tetrahed. Lett., 1969, 59:5185~5188. http://europepmc.org/abstract/med/4391762
-
[2]
A F Fitzpatrick, A M Wynalda. Biochemistry, 1981, 20(21):6129~6134. http://www.ncbi.nlm.nih.gov/pubmed/7306500
-
[3]
M T Ehrman, J D Barlow, J P Hylands. J. Chem. Inf. Model., 2007, 47(2):264~278. http://europepmc.org/abstract/med/17381165
-
[4]
A F Fitzpatrick. Anal. Chem., 1978, 50(1):47~52. doi: 10.1021/ac50023a017
-
[5]
F J Shimitz, T Yasumoto. J. Nat. Prod., 1991, 54(6):1469~1490.
-
[6]
J R Andersen, J D Faulkner, H C He et al. J. Am. Chem. Soc., 1985, 107(19):5492~5495. doi: 10.1021/ja00305a027
-
[7]
N Lindquist, W Fenical, V Duyne et al. J. Org. Chem., 1988, 53(19):4570~4574. doi: 10.1021/jo00254a029
-
[8]
W D Inman, M O'Neill-Johnson. J. Am. Chem. Soc., 1990, 112(1):1~4. doi: 10.1021/ja00157a001
-
[9]
R R West, C L Mayne, C M Ireland. Tetrahed. Lett., 1990, 31:3271~3274.
-
[10]
C J Smith, D A Venables, C Hopmann et al. J. Nat. Prod., 1997, 60:1048~1050. http://europepmc.org/abstract/MED/9358648
-
[11]
P W Ford, B S Davidson. J. Nat. Prod., 1997, 60:1051~1053. http://europepmc.org/abstract/MED/9358649
-
[12]
P Ralifo, L Sanchez, N C Gassne et al. J. Org. Chem., 2007, 70:95~99. http://europepmc.org/articles/PMC1839848
-
[13]
J Kim, E O Pordesimo, S I Toth et al. J. Nat. Prod., 1993, 56:1813~1816. http://europepmc.org/abstract/MED/8277319
-
[14]
V Sharma, P C Sharma, V Kumar. J. Adv. Res., 2015, 6:63~71. http://europepmc.org/abstract/med/25685544
-
[15]
F J Schmitz, S K Agarwal, S P Gunasekera et al. J. Am. Chem. Soc., 1983, 105:4835~4836.
-
[16]
A Plubrukarn, B S Davidson. J. Org. Chem., 1998, 63:1657~1659. doi: 10.1021/jo9719721
-
[17]
G Koren-Goldshlager, M Aknin, E M Gaydou et al. J. Org. Chem., 1998, 63:4601~4603.
-
[18]
G Koren-Goldshlager, M Aknin, E M Gaydou et al. J. Nat. Prod., 2000, 63:830~831.
-
[19]
A Rudi, Y Kashman. J. Org. Chem., 1989, 54:5331~5337.
-
[20]
D C Jenkins, R Armitage, T S Carrington. Zeitschrift für Parasitenkunde, 1980, 63:261~269. http://europepmc.org/abstract/MED/7434874
-
[21]
Z Thale, T Johnson, K Tenney et al. J. Org. Chem., 2002, 67:9384~9391. http://europepmc.org/abstract/MED/12492342
-
[22]
文艳. 中国渔业经济, 2006, 3: 43~45. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=zyjj200603012&dbname=CJFD&dbcode=CJFQ
-
[23]
Y Kitahara, T Mizuno, A Kubo. Tetrahedron, 2004, 60:4283~4288. http://www.sciencedirect.com/science/article/pii/S0040402004004612
-
[24]
G Conte, A J Bortoluzzi, H Gallardo. Synthesis, 2006, 23:3945~3947. doi: 10.1055/s-2006-950323
-
[25]
J Bolger, A Gourdon, E Ishow et al. Chem. Commun., 1995, 17:1799~1800. http://pubs.rsc.org/en/content/articlepdf/1995/c3/c39950001799
-
[26]
A F M Motiur Rahman, Y Kwon, Y Jahng et al. Heterocycles, 2005, 65:2777.
-
[27]
L Guo, B Qiu, G Chen. Anal. Chim. Acta, 2007, 588:123~130. http://www.sciencedirect.com/science/article/pii/S0003267007002632
-
[28]
C Metallinos, F B Barrett, Y Wang et al. Tetrahedron, 2006, 62:11145~11157. http://www.sciencedirect.com/science/article/pii/S0040402006014736
-
[29]
R Elvira, Y Z Hu, F Bouvier et al. Inorg. Chem., 2001, 40:2541~2546.
-
[30]
Y Z Hu, Q Xiang, R P Thummel. Inorg. Chem., 2002, 41:3423~3428. doi: 10.1021/ic011126f
-
[31]
S Gladiali, G Chelucci, M S Mudadu et al. J. Org. Chem., 2001, 66:400~405.
-
[32]
D V Kravchenk, V V Kysil, A P Ilyn. Bioorg. Med. Chem. Lett., 2005, 15:1841~1845. http://www.sciencedirect.com/science/article/pii/S0960894X05001964
-
[33]
D V Kravchenk, V M Kysil, S E Tkachenk et al. Eur. J. Med. Chem., 2005, 40:1377~1383.
-
[34]
E Campaigne, J H Hutchinson. J. Hetercyc. Chem., 1970, 7:655~659.
-
[35]
J Parrick, A Yahya, A S Ijaz et al. J. Chem. Soc., Perkin Transac., 1989, 11:2009~2015.
-
[36]
Y Kitahara, F Tamura, A Kubo. Tetrahed. Lett., 1997, 38:4441~4442.
-
[37]
E Delfourne, F Darro, N Bontemps-Subielos et al. J. Med. Chem., 2001, 44:3275~3282.
-
[38]
M Ȃlvarez, L Feliu, W Ajanaa et al. Tetrahedron, 2000, 56:3703~3708.
-
[39]
刘殿卿, 李德鹏, 由业诚等. 化学研究与应用, 2008, 12: 1613~1616. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=hxyj200812017&dbname=CJFD&dbcode=CJFQ
-
[40]
于小松, 刘殿卿, 由业诚等. 国际网上化学学报, 2008, 10(8): 39. http://kns.cnki.net/KCMS/detail/detail.aspx?filename=hxyj200812017&dbname=CJFD&dbcode=CJFQ
-
[41]
K Tatsuta, S Hosokawa. Chem. Rev., 2005, 105:4707~4729.
-
[1]
-

图 1 Plakortis quasiamphiaster采集实物图
Figure 1 Underwater (left) and abovewater (right) pictures of Plakortis quasiamphiasterb
-


 下载:
下载:





 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 9
- 文章访问数: 2078
- HTML全文浏览量: 198
 下载:
下载:
