Citation:
Zijian Zhang, Fangxin Du, Lijia Liu, Yixiang Sun, Jie Xue, Hanshen Xin. 1-Azaazulene constructs: Overcoming the weak fluorescence of azulene derivatives via S1-S0 transitions[J]. Chinese Chemical Letters,
2026, 37(6): 111987.
doi:
10.1016/j.cclet.2025.111987
1-Azaazulene constructs: Overcoming the weak fluorescence of azulene derivatives via S1-S0 transitions
English
1-Azaazulene constructs: Overcoming the weak fluorescence of azulene derivatives via S1-S0 transitions
School of Microelectronics, Shanghai Engineering Research Center for Integrated Circuits and Advanced Display Materials, Shanghai University, Shanghai 201800, China
inhanshen@shu.edu.cn (H. Xin). 1 These authors contributed equally to this work.
Received Date:
03 June 2025 Accepted Date:
20 October 2025 Revised Date:
14 October 2025 Available Online:
15 June 2026
Abstract:
Azulene derivatives are promising for optoelectronics but suffer from weak fluorescence due to anti-Kasha behavior. This study designed and synthesized four asymmetric 1-azaazulene-based BF2 complexes (Ph-NN, Ph-ON, Ca-NN, Py-NN) to explore their optoelectronic potential. Unlike azulene derivatives, which predominantly exhibit S2→S0 emission, 1-azaazulene constructs demonstrate effective S1-S0 transitions, resulting in enhanced fluorescence. Py-NN, incorporating a pyrrole group, achieved a remarkable photoluminescence quantum yield (PLQY) of 19.3% at 560 nm, attributed to enhanced HOMO-LUMO orbital overlap with an oscillator strength (f) of 0.4973, resulting in a high radiative rate (kr = 9.51 × 107 s−1) and suppressed non-radiative decay (knr = 0.40 × 109 s−1). OLEDs employing Py-NN as an emitter exhibited orange electroluminescence at 565 nm with a maximum external quantum efficiency (EQE) of 3.0%. The results demonstrate that 1-azaazulene can serve as a versatile platform for developing efficient fluorescent materials with applications in optoelectronics.
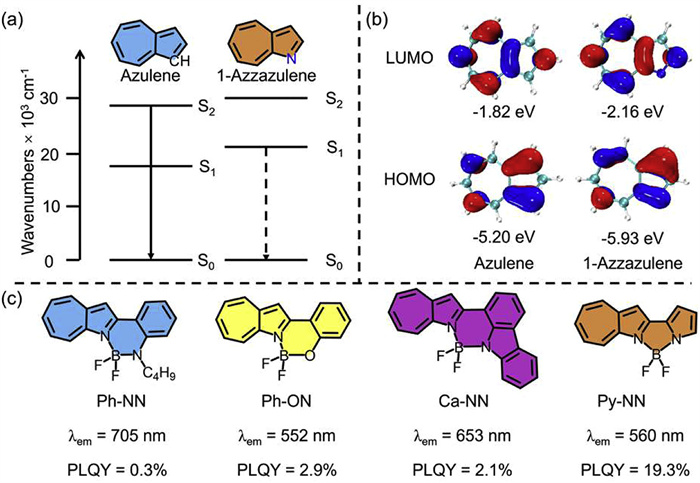
Azulene has emerged as a promising candidate for constructing advanced organic optoelectronic materials due to its unique optoelectronic properties [1-6]. In recent years, azulene-based materials have been extensively investigated in organic field-effect transistors (OFETs), solar cells, and other related applications [7-14]. As illustrated in Figs. 1a and b, the asymmetric frontier molecular orbitals (FMO) of azulene reduce electron repulsion between unpaired electrons, resulting in a narrow energy gap between its ground state (S0) and first excited state (S1). This reduced energy gap leads to a visible-range absorption band (S0 → S1 transition), endowing azulene with its characteristic blue color [15-17]. Furthermore, the significant energy difference between S1 and S2 (∆E(S2-S1) > 10,000 cm−1) coupled with orbital asymmetry slows the non-radiative S2 → S1 transition while enhancing the radiative S2 → S0 pathway. This gives rise to the anti-Kasha fluorescence behavior observed in azulene [18,19]. However, this unique photophysical property also confines the fluorescence of azulene and its derivatives to the short-wavelength region with inherently weak emission intensities [20-25]. For example, the reported azulene-based BN-heteroaromatics exhibit photoluminescence quantum yield (PLQY) of less than 0.5% (Fig. S1a in Supporting information) [24].
Figure 1
Figure 1.
(a) Chemical structures and schematic diagrams of radiation transitions for azulene and 1-azaazulene. (b) Frontier molecular orbitals and their energetics of azulene and 1-azaazulene. (c) Chemical structures, emission peaks, and photoluminescence quantum yields of the 1-azaazulene-based BF2 complex.
As illustrated in Fig. 1a, replacement of the CH moiety at the 1-position of azulene with a N atom yields its heteroanalog, 1-azaazulene. Density functional theory (DFT) calculations demonstrate that N substitution profoundly alters the electronic structure and FMO energetics (Fig. 1b). Compared to pristine azulene (HOMO/LUMO = −5.20/−1.82 eV, Egap = 3.38 eV), 1-azaazulene exhibits stabilized HOMO (−5.93 eV) and LUMO (−2.16 eV) levels, with the bandgap widening to 3.77 eV due to a more pronounced stabilization of the HOMO level. Time-dependent DFT (TD-DFT) calculations reveal that while 1-azaazulene retains asymmetric frontier orbitals, the S0-S1 energy gap becomes substantially wider than that of the azulene. Notably, the reduced S2-S1 energy separation accelerates the non-radiative S2 → S1 conversion compared to azulene, effectively suppressing the anti-Kasha S2 → S0 radiative transition. Such changes in electronic structure and FMO energetics render 1-azaazulene a promising scaffold for designing Kasha-compliant fluorescent materials with red-shifted emission profiles compared to its quinoline isomer (Fig. S2 in Supporting information) [26-29].
Boron difluoride (BF2) complexes represent a prominent class of luminescent materials [30-35]. The lone pair electrons on the N atom of 1-azaazulene enable effective coordination, making it an ideal ligand for constructing BF2 complexes [36-40]. In this study, four asymmetric 1-azaazulene-based BF2 complexes (Ph-NN, Ph-ON, Ca-NN, and Py-NN, Fig. 1c) were designed, synthesized, and systematically investigated for their photophysical properties. The luminescence wavelength and efficiency of these complexes were modulated by structural variations in the ancillary coordination groups. In contrast to the anti-Kasha fluorescence observed in azulene derivatives, all four 1-azaazulene constructs display S1 → S0 emission fluorescence. Ph-NN exhibits an emission peak at 705 nm, while Py-NN achieves a PLQY of 19.3%. Furthermore, the organic light-emitting diode (OLED) fabricated with Py-NN as the emitter via a sensitization strategy delivers a maximum external quantum efficiency (EQEmax) of 3%.
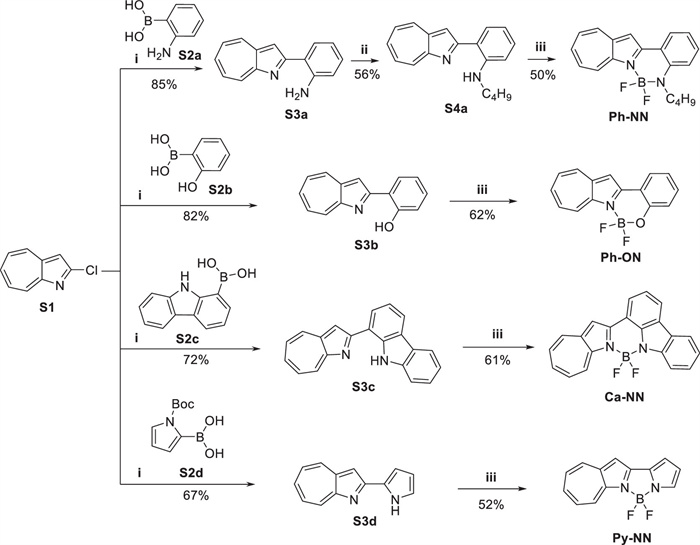
The synthetic routes for Ph-NN, Ph-ON, Ca-NN, and Py-NN are illustrated in Scheme 1. First, 2-chloro-1-azaazulene (S1) was synthesized according to the literature procedure [41,42]. Subsequently, Suzuki-Miyaura coupling reactions between S1 and various arylboronic acids S2a-S2d were performed, affording intermediates S3a-S3d in 67%-85% yields. Notably, for reaction of S1 and S2d, treatment with sodium methoxide during workup enabled in situ deprotection of the Boc group, directly affording intermediate S3d. To prevent potential interference of the amino group during BF2 complex formation, S3a was substituted with an alkyl chain to obtain intermediate S4a. Finally, exposure of intermediates S4a and S3b-d to excess BF3·Et2O in toluene, in the presence of TEA/DIPEA, afforded 1-azaazulene-based BF2 complexes Ph-NN, Ph-ON, Ca-NN, and Py-NN with 50%-62% yields. These compounds were characterized by nuclear magnetic resonance (NMR) spectroscopy and high-resolution mass spectrometry (HRMS). The full characterization data can be found in Supporting information.
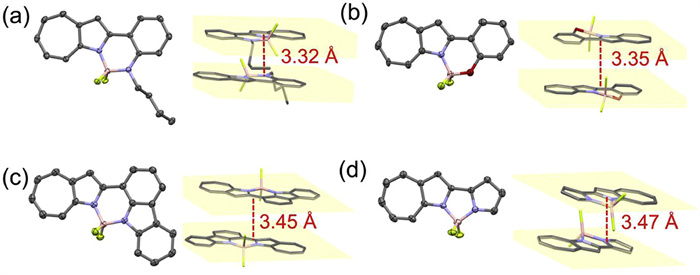
The structures of Ph-NN, Ph-ON, Ca-NN, and Py-NN were further confirmed through X-ray single crystal analysis (Fig. 2, Tables S1-S4 in Supporting information). The single-crystal structures reveal that all four compounds possess highly planar backbones, with the largest dihedral angles in the fused rings being less than 4.95° (Fig. S3 in Supporting information). The crystal packing diagrams in Fig. 2 and Figs. S4-S7 (Supporting information) demonstrate that in Ph-NN, Ph-ON, Ca-NN, and Py-NN, adjacent molecules are arranged through antiparallel stacking of their conjugated backbones. The stacking distances, ranging from 3.32 Å to 3.47 Å, suggest that all four molecules have strong π-π interactions. These pronounced π-π interactions lead to a decrease in the PLQY of the thin films due to aggregation-caused quenching (ACQ) effects (Table S5 in Supporting information). As shown in Fig. S3, in the four BF2 complexes, the average bond lengths (1.38–1.42 Å) of the 1-azaazulene and its connected aromatic rings are close to the bond length of aromatic C=C bonds, indicating that the ligand backbones maintain good aromaticity. The NICS(1)zz values (Fig. S8 in Supporting information) and the anisotropy of the induced current density (AICD) plots (Fig. S9 in Supporting information) further confirm the aromatic characteristics of the 1-azaazulene-based ligand backbones. Notably, the N→B coordination bonds in these four molecules (1.55–1.58 Å) are significantly shorter than those reported in asymmetric BF2 complexes [43,44]. The short N→B bond lengths reflect strong dative interactions between nitrogen and boron, enhancing the complex’s stability and ensuring the integrity of the fluorescent materials under various conditions.
Figure 2
Figure 2.
Crystal structures and packing diagrams of (a) Ph-NN, (b) Ph-ON, (c) Ca-NN, and (d) Py-NN.
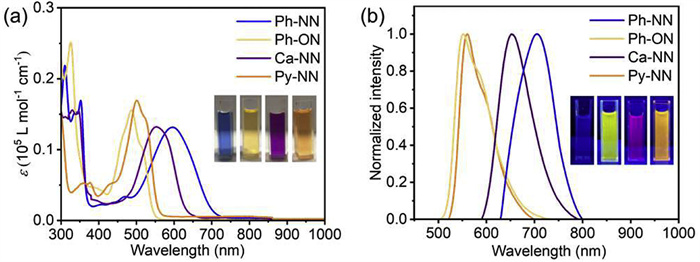
To investigate the photophysical properties of Ph-NN, Ph-ON, Ca-NN, and Py-NN, we performed absorption spectroscopy, steady-state, and time-resolved emission spectroscopy under oxygen-free conditions at a concentration of 1 × 10−5 mol/L (Table 1). As shown in Fig. 3a, all four molecules exhibit intense absorption bands in the 300–400 nm range, primarily attributed to S0 → S2 transitions of 1-azaazulene units. However, they exhibit distinct absorption differences in the long-wavelength region. Ph-ON and Py-NN display maximum absorption peaks at 487 and 501 nm, respectively, with pronounced shoulder peaks, indicating dominant localized excitations. In contrast, Ph-NN and Ca-NN show significantly red-shifted absorption at 597 and 555 nm, accompanied by broader absorption bands lacking distinct shoulders, which suggests excitations with pronounced charge-transfer (CT) characteristics. Notably, these absorption bands display significant redshifts compared to analogous quinoline derivatives [43,45,46], which may originate from the asymmetric electron distribution and narrower FMO energy gaps of the 1-azaazulene core. Remarkably, the molar absorption coefficients for the S0 → S1 transitions of these compounds (ε > 104 L mol−1 cm−1) are substantially higher than those of typical azulene-based molecules (ε < 104 L mol−1 cm−1).
Table 1
Table 1.
Summarized key photophysical properties of compounds Ph-NN, Ph-ON, Ca-NN, and Py-NN.
a Meassured in oxygen-free toluene solution (1 × 10−5 mol/L) at room temperature; b Radiative rate constant is calculated as kr =ΦPL/τ; c Non-radiative rate constant is calculated as knr = (1 − ΦPL)/τ.
Figure 3
Figure 3.
(a) UV–vis absorption and (b) emission of compounds Ph-NN, Ph-ON, Ca-NN, and Py-NN (10−5 mol/L in oxygen-free toluene) with pictures inserted.
As depicted in Fig. 3b, Ph-NN exhibits an S1 → S0 emission peak at 705 nm, a rare feature among small-molecule BF2 complexes, with a Stokes shift (S) of 108 nm. However, its PLQY is only 0.3%, likely due to ultrafast nonradiative decay. Time-resolved emission spectroscopy (Fig. S10a in Supporting information) reveals an exceptionally short luminescence lifetime (τ) of 0.18 ns for Ph-NN, yielding a radiative transition rate (kr) of 1.67 × 107 s−1 and a nonradiative rate (knr) of 5.54 × 109 s−1 (Table 1). In contrast, Ph-ON and Ca-NN display blue-shifted emission peaks at 552 nm (S = 74 nm) and 653 nm (S = 98 nm), with improved PLQYs of 2.9% and 2.1%, respectively. Their luminescence lifetimes increase to 0.84 ns (Ph-ON) and 0.59 ns (Ca-NN), corresponding to kr/knr values of 3.45 × 107 s−1/1.16 × 109 s−1 and 3.56 × 107 s−1/1.66 × 109 s−1 (Figs. S10b and c in Supporting information). Their enhanced PLQYs arise from both increased kr and reduced knr. Significantly, Py-NN demonstrates the highest luminescence efficiency among the four 1-azaazulene-based BF2 complexes, exhibiting an emission peak at 560 nm (S = 59 nm) with a PLQY of 19.3%. The relatively small Stokes shift and high emission efficiency suggest dominant localized-state emission in Py-NN. Time-resolved photoluminescence spectrum (Fig. S10d in Supporting information) confirms a prolonged luminescence lifetime of 2.03 ns, corresponding to kr = 9.51 × 107 s−1 and knr = 0.40 × 109 s−1. These results indicate that by altering the structure of the groups attached to the 1-azaazulene, it is possible to tune the photophysical properties including absorption spectrum, emission spectrum, and emission efficiency of 1-azaazulene-based BF2 complexes.
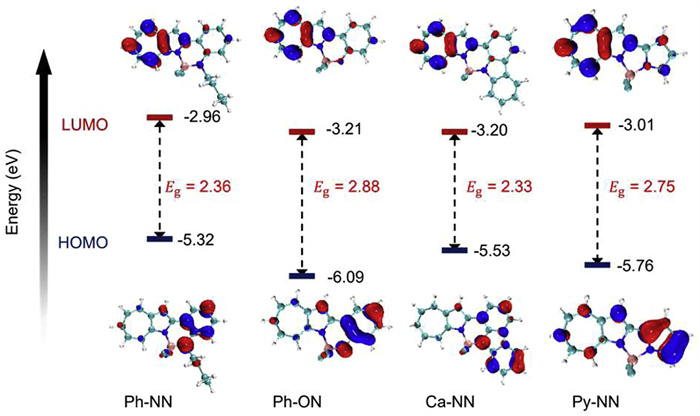
DFT calculations at the B3LYP/6–31G+(d,p) level were employed to investigate the electronic properties of the four 1-azaazulene-based BF2 complexes, with the calculated data summarized in Table S6 (Supporting information). As illustrated in Fig. 4, the FMO of Ph-NN, Ph-ON, Ca-NN, and Py-NN exhibit significantly uneven electron density distributions, a notable contrast to most BF2 complexes [47-49]. Specifically, the LUMO electron density is primarily distributed over the 1-azaazulene unit, while the HOMO electron density is mainly localized on the aniline, phenol, carbazole, and pyrrole moieties. For Ph-ON and Py-NN, especially the latter, the HOMO electron density also has a small distribution on the 1-azaazulene. This pronounced asymmetry in molecular orbital distribution suggests a propensity for charge-transfer (CT) transition absorption in these compounds, while Ph-ON and Py-NN may also exhibit localized transition absorption. As shown in Fig. 4, the calculations indicate that by altering the structure of the group connected to the 1-azaazulene, the FMO energetics and band gap of the 1-azaazulene-based BF2 complexes can be effectively modulated, thereby tuning the absorption and emission wavelengths of the molecules.
Figure 4
Figure 4.
Frontier molecular orbitals, and their energetics of compounds Ph-NN, Ph-ON, Ca-NN, and Py-NN, obtained by DFT and TD-DFT calculations.
Since the B3LYP functional may underestimate the energy of charge-transfer electronic states [50], the long-range corrected M062X functional was employed to study the excited states of 1-azaazulene-based BF2 complexes using the 6–31G(d) basis set. TD-DFT calculations show that the oscillator strengths (f) for the S0 → S1 transition of Ph-NN, Ph-ON, Ca-NN, and Py-NN are 0.3299, 0.3009, 0.3458, and 0.4973, respectively. The increased f could originate from the superior hole-electron overlap of S1 state of Py-NN compared with the other three molecules. This indicates that Py-NN likely exhibits accelerated radiative decay rate (kr). In addition, Py-NN demonstrated a relatively large energy gap (ΔE = 2.75 eV) and smallest reorganization energy (λM = 3942 cm−1), which is conducive to decelerating nonradiative decay rate (see Supporting information for details). Accordingly, Py-NN with large kr and small knr likely exhibits high PLQY, which agrees well with the experimental results. In the electron-hole overlap maps of the S1 excited state and atomic transition density matrix (TDM) heat maps for the S0 → S1 transition (Fig. S10e in Supporting information), Ph-ON and Py-NN show substantial electron-hole overlap, primarily localized on the 1-azaazulene unit. This localization is favorable for the formation of localized excited states (LE). Additionally, the maps demonstrate that the electronic excitation process is chiefly focused on electron transfer among the atoms within the 1-azaazulene unit.
Cyclic voltammetry (CV) and differential pulse voltammetry (DPV) tests were performed on Ph-NN, Ph-ON, Ca-NN, and Py-NN, as shown in Figs. S11a-d and Table S8 (Supporting information). Electrochemical calculations determined the HOMO/LUMO levels of Ph-NN, Ph-ON, Ca-NN, and Py-NN to be −5.39/−3.61 eV, −6.39/−3.57 eV, −5.42/−3.70 eV, and −5.73/−3.63 eV, respectively. Overall, the FMO energy levels obtained from electrochemical measurements, particularly the HOMO levels, were consistent with the theoretical computational trends. Additionally, thermogravimetric analysis (TGA) was conducted and the results indicated that the thermal decomposition temperatures (Td) for Ph-NN, Ph-ON, Ca-NN, and Py-NN were 273, 246, 371, and 428 ℃ (Fig. S12 in Supporting information), respectively, demonstrating good thermal stability across all compounds.
Owing to the moderate PLQY and superior thermal stability of Py-NN, OLED devices were fabricated to evaluate the electroluminescent performance of 1-azaazulene-based BF2 complexes with multilayer configurations of ITO/HATCN (5 nm, hole injection layer)/NPB (40 nm, hole-transport layer)/TCTA (10 nm, exciton-blocking layer)/TPBi: 20 wt% thermally activated delayed fluorescence sensitizer 4CZIPN and 1 wt% Py-NN (30 nm, emissive layer)/TPBi (25 nm, electron-transport layer)/LiF (1 nm, electron-injection layer)/Al (150 nm) (Fig. 5a and Fig. S12). As shown in Fig. 5b, the device exhibits strong orange emission with the corresponding electroluminescence peak value (λem) located at 565 nm, which is close to the photoluminescnece peak of Py-NN. And the OLED exhibited an EQEmax of 3.0% and a turn-on voltage of 5.2 V.
Figure 5
Figure 5.
(a) OLED device architecture. (b) Electroluminescent emission spectrum at 6 V. (c) The J–V–L (current density–voltage–luminance) characteristics. (d) The EQE–J (external quantum efficiency–current density) relationship.
In summary, this study successfully designed and synthesized four asymmetric 1-azaazulene-based BF2 complexes (Ph-NN, Ph-ON, Ca-NN, and Py-NN) to explore their optoelectronic properties. By modifying ancillary coordination groups, the FMO energetics and band gaps of these complexes were effectively tuned, allowing adjustments in their absorption and emission properties. All four 1-azaazulene constructs exhibit S1 → S0 emission fluorescence, in contrast to azulene derivatives, which typically display anti-Kasha fluorescence and low PLQY. Notably, Py-NN demonstrated superior performance, achieving a PLQY of 19.3% with an emission peak at 560 nm. This enhancement is attributed to improved HOMO-LUMO overlap and suppressed nonradiative decay pathways. OLEDs incorporating Py-NN as an emissive dopant exhibited a maximum EQE of 3%, underscoring its electroluminescent potential. These findings demonstrate that 1-azaazulene serves as a promising scaffold for developing fluorescent materials, overcoming the inherent limitations of traditional azulene derivatives and advancing its potential applications in optoelectronic devices. Moreover, 1-azaazulene constructs possess smaller band gaps compared to traditional fluorescent materials with similar-sized π-conjugated systems. This characteristic facilitates the development of long-wavelength emission fluorescent materials, which are desirable for applications requiring deep-red or near-infrared emission.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statement
Zijian Zhang: Writing – original draft, Formal analysis, Data curation. Fangxin Du: Writing – original draft, Methodology, Formal analysis. Lijia Liu: Formal analysis, Data curation. Yixiang Sun: Formal analysis. Jie Xue: Writing – review & editing, Methodology, Investigation. Hanshen Xin: Writing – review & editing, Writing – original draft, Project administration, Investigation, Funding acquisition, Conceptualization.
Acknowledgments
This research was financially supported by the Natural Science Foundation of Shanghai Municipality (No. 25ZR1402168), the National Natural Science Foundation of China (No. 22101170) and Shanghai University. We thank Wen-wen Zhang and Jinheng Pan (Mass Spectrometry & Metabolomics Core Facility of Westlake University) for MS analysis.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.111987.
[1]
A. Ong, T. Tao, Q. Jiang, et al., Angew. Chem. Int. Ed. 61 (2022) e202209286. doi: 10.1002/anie.202209286
Figure 1
(a) Chemical structures and schematic diagrams of radiation transitions for azulene and 1-azaazulene. (b) Frontier molecular orbitals and their energetics of azulene and 1-azaazulene. (c) Chemical structures, emission peaks, and photoluminescence quantum yields of the 1-azaazulene-based BF2 complex.
Figure 3
(a) UV–vis absorption and (b) emission of compounds Ph-NN, Ph-ON, Ca-NN, and Py-NN (10−5 mol/L in oxygen-free toluene) with pictures inserted.
Table 1.
Summarized key photophysical properties of compounds Ph-NN, Ph-ON, Ca-NN, and Py-NN.
Comp.
λabs (nm)a
λem (nm)a
ΦPL (%)a
τ (ns)a
kr (107 s−1)b
knr (109 s−1)c
Ph-NN
597
705
0.3
0.18
1.67
5.54
Ph-ON
487
552
2.9
0.84
3.45
1.16
Ca-NN
555
653
2.1
0.59
3.56
1.66
Py-NN
501
560
19.3
2.03
9.51
0.40
a Meassured in oxygen-free toluene solution (1 × 10−5 mol/L) at room temperature; b Radiative rate constant is calculated as kr =ΦPL/τ; c Non-radiative rate constant is calculated as knr = (1 − ΦPL)/τ.

 DownLoad:
DownLoad:


 下载:
下载:







 下载:
下载:
