Figure 1.
The theoretical insights, reaction mechanism, characterization methods and atom/support/reactants effects for determining the electronic structure of single atoms.

The development of "single-atom catalysis" provides a brand-new concept for constructing high-efficiency surface active sites from the atomic scale, which is expected to maximize the utilization of atoms and effectively reduce the demand of precious metal elements in catalysts [1]. The rational design of single atom catalysts (SACs) and the development of controllable synthesis have extended the research in catalysis area. At present, most of the studies about single-atom catalysis focus on the optimization of catalytic performance [2-4]. Adjusting the geometric configuration and coordination environment of single-atom active sites can tune the catalytic reaction path and thus improve the catalytic activity. Although some catalytic performance progress of SACs has been made, gaining deeper insight into the bonding energy and further understanding the catalytic mechanism of single atom catalysis is an important frontier in catalysis research.
The catalytic activity and selectivity of SACs highly depend on the local coordination environment of the metal center, that is, the electronic and geometric interaction between a single atom and the carrier. In catalytic reactions, the chemical bonding energy of the SACs is one of the important factors affecting the activity of the catalyst. Low bonding energy makes the SACs more susceptible to react with reactants, promoting the adsorption and activation of reactants, thereby reducing the activation energy of the reaction. The bonding energy has various implications for catalytic reactions, influencing aspects such as the rate, selectivity, stability, and mechanism of SAC related reactions. In addition, the stability of the SACs is closely related to its surface bonding energy. The strong bonding energy between a single metal atom and its neighboring coordinating atoms makes the catalysts more stable, thereby improving the durability of the catalyst. Therefore, it is necessary to fully consider the characteristics of the bonding energy of the SACs.
The bonding energy between a single metal atom and its neighboring coordinating atoms, as well as the metal-support interaction are the main factors which determine the charge density and distribution of the metal sites, thereby affecting the overall catalytic performance of SACs. With the formation of strong interaction between a single metal atom and the support, an obvious charge delocalization and electron redistribution arising from the electron transfer between the metal and the support can be detected. Therefore, some of the metal atoms in SACs are positively charged and exhibit a high valence state like cation [5-7]. The tuned electronic structure of single atom sites plays an important role for their unique catalytic activities in specific catalytic reactions. In addition, the active site of a single atom will undergo reconstruction of the atomic configuration, accompanied by a change in valence during the catalytic reactions. This means that the interaction between a single metal atom and the support will self-regulate, thereby promoting the progress of the catalytic reaction. In-situ real-time characterization of the electronic structure provides strong evidence to understand the nature of single-atoms during reaction [8-12]. Although there have been several reviews summarizing the development of SACs, most of them focused on the synthesis and applications. Almost no reviews have focused on the sights of atomic bonding environment and real-time electronic structure of SACs during catalytic reactions. Probing the response behavior of the electronic structure of SACs is of great significance for understanding the nature of single-atom catalysis and discovering new catalytic phenomenon.
This review describes recent progress on the understanding the electronic structures of single atom sites and the bonding energy between the single atoms, supports and reactants (Fig. 1). Firstly, we briefly introduce the theoretical insights in single atom catalysis, such as the density functional theory (DFT) for determining the electronic structure of SACs, the electron transfer simulation during catalytic reactions. Then three characterization methods for testing the electronic structure and coordination environments are introduced. The detailed information about the atomic bonding energy in SACs are described into the bonding energy between SACs and substrates; the bonding energy between neighboring metal atoms; the bonding energy between SACs and reactants during reactions.
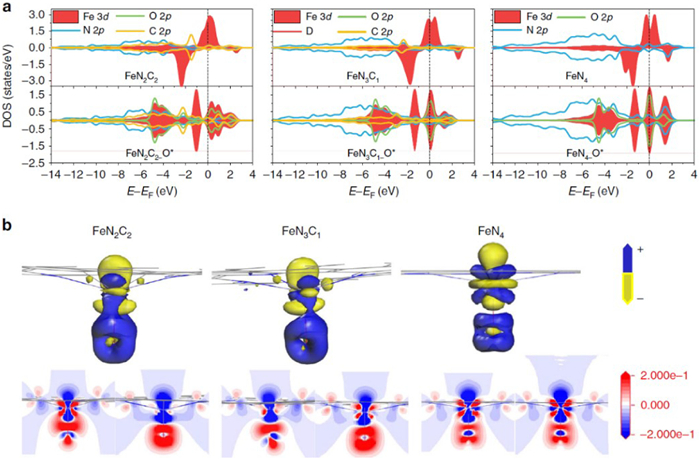
Because the atoms are isolated located on the substrates in SACs, the DFT simulation models can be accurately fabricated to analyze the electronic structure of single atoms. The electronic structure analysis of single atoms could provide useful information for understanding their catalytic reaction mechanisms and providing principles for design future single atoms. In many studies, density of states (DOS) calculations is employed to speculate the electron dispersion at near the Fermi level for the SAC systems. For example, Chen and co-workers calculated the DOS for the Fe-N2C2, Fe-N3C1, Fe-N4 centers and oxygen in Fe=O species (Fig. 2) [13]. The DOS results show that the C 2p orbitals of Fe-N2C2 and Fe-N3C1 are close to the Fermi energy level, suggesting that the stronger interactions between two- and three-N doped graphene with Fe atom. The calculated Hirshfeld charge population of O atoms in Fe=O show that the bonding strength between Fe-N4 and O atom is the weakest, while the charge transfer in it is the strongest. Sun and co-workers found that when Pt single atoms were bonded with N, the Pt 5d-orbitals and N 2p-orbitals mix at around the Fermi level [14]. The calculated Bader charges revealed that there is an electron transfer from Pt to N in the sample of Pt1nullNC. During the hydrogen evolution reaction, the 5d orbitals of Pt atoms interact strongly with the 1s orbital of the H atoms, leading to electron pairing and hydride formation. When compared with Pt clusters, more electron was found transfer from Pt single atom to H, indicating that the single Pt atoms on the substrates become nonmetallic through the donation of electrons to both the substrate and H atoms.
In addition to the coordination with non-metallic elements of C, N or O, the single atoms can also be deposited onto metal surfaces and forming the single atom alloy (SAA) structure. The electronic structures of SAA are also analyzed by the DFT DOS simulation. Zhang et al. prepared the isolated Pt on Au surface by a facile colloidal method [15]. The DFT DOS results predict a drastic reduction of intensity near the Fermi level for the SAAs relative to the pure Pt surface. In addition, the SAAs can exhibit a free-atom-like d states of the minority element resulting from the weak wave function mixing between minority and majority elements. Greiner and Jones investigated the electronic structure of Cu in Cu/Ag SAAs [16]. The width of the Cu d band of 0.5 eV in AgCu is much smaller than that of bulk Cu. The narrow d band effectively increases the interaction strength with the O 2p levels, which caused the involvement of 2px and 2py orbitals and the π bonds between O and Cu.
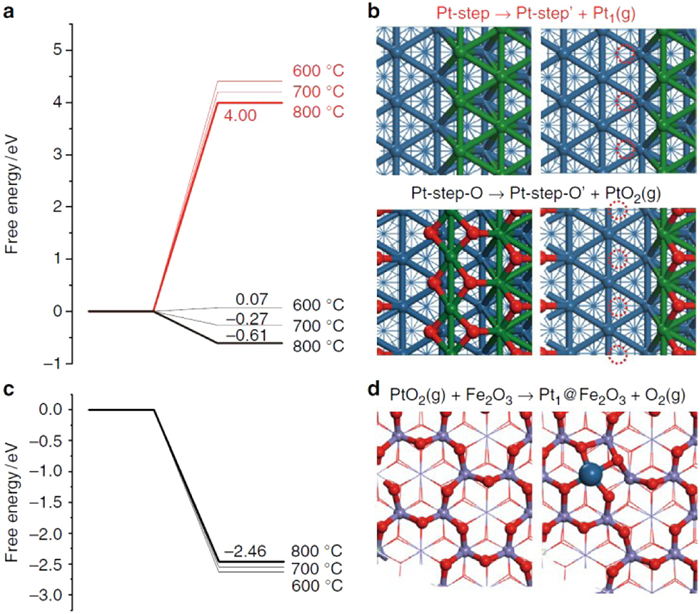
The highly dispersed single atom catalysts have the atom utilization up to 100%, which exhibit unique performance in catalytic activity and selectivity. However, the extremely high surface energy results in the easy aggregation and stability challenge of single atoms. Recently, the increasingly attention has been received in the field of how to fabricate the high stable SACs. The creation of strong metal–support interaction (SMSI) can effectively stabilize single atoms on their substrate. Therefore, the both the experimental and theoretical simulation research are very necessary in deep understanding the interaction between single atoms and supports. The electron transfers between the single atoms and the support can alter the electronic structure of the metal atoms, thus leading to a crucial effect on the catalytic activity and stability in heterogeneous catalysis. Zhang and Li report high concentrations of thermally stable Pt single atoms with a strong metal–support interaction on the FeOx support [17]. The DFT calculation results show that the stable species is PtO2 with evaporation energy of only −0.61 eV from a Pt(221) step at 800 ℃; while the evaporation energy of Pt1 is as high as 4.00 eV which is strongly disfavoured (Figs. 3a and b). In addition, the dissociative adsorption of PtO2 on Fe2O3(0001) surface (with an oxygen terminated surface) is calculated to be −2.46 eV, which is highly exothermic at 800 ℃ (Figs. 3c and d). The computational modelling concluded the formation of Pt atoms coordinate with four surface oxygen atoms in a distorted square geometry with an average Pt–O length of 1.94 Å. The total free energy change for the formation of Fe2O3 stabilized isolated Pt atoms from Pt NPs is −3.07 eV per Pt atom at 800 ℃, confirming the SMSI of Pt atoms with Fe and O atoms of the surface.
Besides, the theoretical calculations can speculate the preferred bonding sites of single atoms. Au single atoms prefer to deposit on the step sites of CeO2 according to the theoretical results [18]. Fabris et al. stabilized the Pt single atoms on CeO2 by surface step decoration [19]. The step edges with intrinsic defects on nanostructured ceria surfaces can effectively stabilize Pt single atoms in the form of planar PtO4 moieties. When Pt are deposited onto doped substrates, the anchor sites which help in immobilizing Pt atoms can also be determined by DFT calculations. Sun and Liu studied the stabilization mechanism of Pt atoms on N-doped graphene by DFT calculations [14]. The adsorption energy of Pt atom directly bond to the N-dopant is calculated to be 5.171 eV, which is much higher than other structures. Moreover, the calculated Bader charge shows that 0.257 e is transferred from the Pt to the N atom in the N-doped graphene substrate in this stable model, confirming the relatively large Pt adsorption energy at this N-anchoring site.
A comprehensive understanding of the atomic bonding energy and intermediates adsorption/desorption energy can help to promote the catalytic performance to a new level for single atom catalysis. Till now, the reaction mechanism for several thermal catalytic reactions including CO oxidation and propane dehydrogenation, are illustrated on different types of single atoms based on the calculation modeling [20-25]. For CO oxidation, three different mechanisms (Mars van Krevelen, Langmuir–Hinshelwood, and Eley–Rideal mechanisms) are raised. In the Mars van Krevelen mechanism, the O lattice of Cu2O in the atomically dispersed Pt1/Cu2O combines with adsorbed CO to form CO2 and this elementary steps the rate determining step during the CO oxidation process [25]. On the CeO2 surface, the Pd single atoms presents in the form of PdO and PdO2. The O of PdO or PdO2 and CO are co-adsorbed on the catalyst with the Langmuir–Hinshelwood mechanism [20]. The Eley–Rideal mechanisms are found on the single atom Ir supported on MgAl2O4 [24]. The adsorption of CO weakens the O binding strength on Ir sites and the adsorbed O2 can dissociate easily to heal the vacancy with a negligible barrier after the creation of open coordination at Ir single atoms. The propane dehydrogenation (PDH) to form propylene is another important catalytic reaction owing to the growing demand for propylene. In the Pd/Cu SAAs, the introduction of a single Pd atom on the Cu cluster can effectively promote the C–H bond activation during PDH. The side reactions can be inhibited because of the desorption of relatively weakly adsorbed H atoms on the inactive Cu surface [22].
In addition to thermal catalysis, the SACs are also widely applied in electrochemical applications. For example, hydrogen evolution reaction (HER) is one of the promising clean routes for the production of hydrogen in the future. Pt single atoms exhibited extremely high mass activity compared with Pt nanoparticle catalysts. DFT calculation results show that one Pt single atom can adsorb with several H atoms, before the formation of H2 on the isolated Pt atom [26,27]. The desorption and hydrogen evolution process is similar as the Tafel reaction (Hads + Hads → H2). Moreover, the non-noble metal SACs are found exhibit comparable oxygen reduction reaction (ORR) activities, which is promising to substitute noble Pt catalysts applied in fuel cells. Chisholm et al. reported the single Nb atom catalysts incorporated in a graphite layer for excellent ORR catalyst [28]. The theoretical simulations indicate that the charge density near the Fermi level of Nb is dense and the barrier for O–O dissociation energy is relatively low. In addition to HER and ORR, the single atoms are also widely applied in electrocatalysis of CO2 reduction reaction. Interestingly, most of the catalysts exhibited good selectivity for CO during the CO2 reduction process [6,29-33]. The DFT calculation results indicate that the formation energy of COOH* is low on the single atom sites, leading to the high selectivity of CO.
X-ray absorption spectroscopy (XAS) have become one necessary characterization method for probe the electronic structure of the single atoms. The X-ray absorption near edge spectroscopy (XANES) and extended X-ray absorption fine structure (EXAFS) spectroscopy are two most commonly used technique for identifying the coordination environment and bonding energy of central metal atoms in the SACs. Firstly, the near-edge absorption energy in XANES curves can detect the chemical valence of the metal single atoms. The comparison of as-prepared SACs with metal foil, metal oxide and precursors can qualitatively conclude the oxidation state of the metal single atom [34,35]. Taking the metal-organic frameworks (MOF) derived Fe single atoms as the example, near-edge absorption energy for Fe single atoms located between Fe2O3 and Fe foil, implying Fe single atoms is in partially oxidation states other than in the metallic property. The positively charged state of Fe SACs indicates that the electrons transfers from Fe to the substrate [36]. The peak location (threshold energy E0) of the first derivative of the XANES spectrum can precisely determine the oxidative state. A positive energy shift in the threshold energy E0 means the surface oxidation in the prepared SACs. For example, the threshold energy of Ni SACs/N—C located between those for the Ni foil and NiO in XANES spectra, suggesting the partly charged electronic structure of Niδ+ (0 < δ < 2) [37]. The peak intensity in XANES spectra can reflect the vacancy and occupied electrons in the orbital. The formation of Pt single atoms on carbon-based catalysts can significantly increase the white line density compared to Pt foils, indicating the presence of high density of unoccupied d orbitals states [38].
In addition to qualitative analysis, the quantitative analysis of XANES spectrum can be applied to identify the coordination environment for some specific metals, such as Au and Pt [39]. The qualitative analysis is based on the fact that the electron counts change in d orbitals is primarily responsible for the difference of the area under the white line. Prof. Sham carefully analyzed the hole counts and changes in the white line areas of Au-Cu alloy by subtracting the absorption spectrum at the L2,3 edges of pure Au from that of the alloys. They found that with the formation of Au-Cu alloy structure, the Au site may have a loss of d charge due to the dilution of Au in Cu. In the elemental table, Pt and Au are the neighbors with nearly full or full 5d bands. Therefore, the Pt XANES peak intensity can be analyzed using the Au metal XANES peaks as the background. Sun and co-workers explored the unoccupied electron density of 5d states in Pt single atoms according to the quantitative analysis method [38]. The Pt single atoms on graphene sample has the higher total unoccupied density of states of Pt 5d character than Pt nanoparticles (Figs. 4a and b) [14]. With the doping of N in graphene, the unoccupied density of states of Pt 5d character further increased, and the vacant d-orbitals of individual atoms play a vital role in enhancing the activity of catalysts towards the HER.
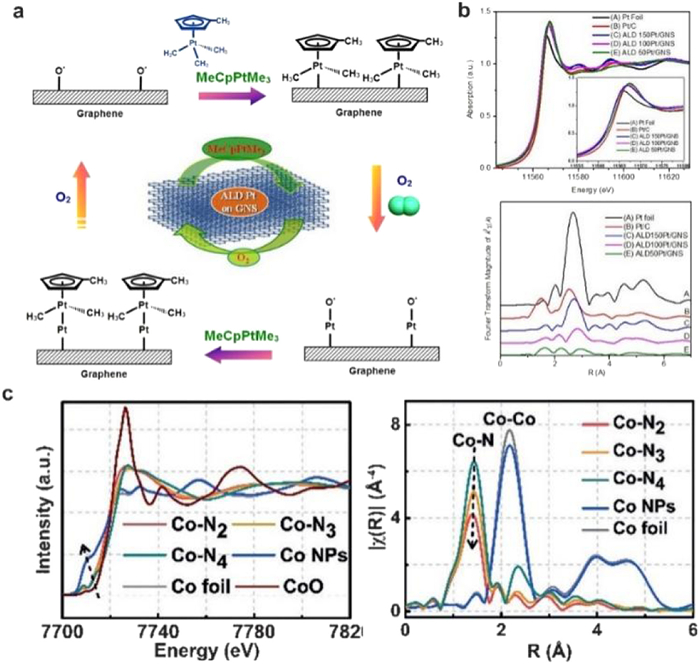
Different from the XANES spectra, the EXAFS spectra are related to the scattering of absorbing atoms and the neighbor atoms next to them. The EXAFS spectra can give the information about the neighboring element atoms around the central absorbing atoms. In addition, the parameters such as the interatomic distance and coordination number can be obtained by fitting the Fourier transform (FT)-EXAFS R space curves. For example, Li et al. constructed Co SACs on the nitrogen-doped porous carbon support by in-situ pyrolysis of Zn/Co bimetallic MOF precursors. The coordination environment of obtained Co sites can be well tuned by adjusting the pyrolysis temperatures in the range of 800–1000 ℃. In the FT-EXAFS spectra R space, the intensity of peak at ca. 1.4 Å reduced gradually with the reduction of the coordination number of Co-N from 4 to 2. The fitting results also indicated the accuracy coordination numbers of Co-N are 4.1, 3.1 and 2.2, respectively, which agrees well with the Co-N4, Co-N3, and Co-N2 coordination structures (Fig. 4c) [29]. The wavelet-transform EXAFS (WT-EXAFS) is another commonly used spectrum. Compared with Fourier transform processing of XAS data, WT-EXAFS has its own characteristics. The wavelet transform uses a finite-length Morlet wavelet as the fundamental wave, replacing the infinitely long sine wave fundamental wave in the Fourier transform. Generally, wavelet transform uses colored planar graphs to display 3D information. In addition to showing the location of peaks, it also uses different colors to represent the height of peaks. This allows researchers to more intuitively understand the interactions and coordination structures between atoms.
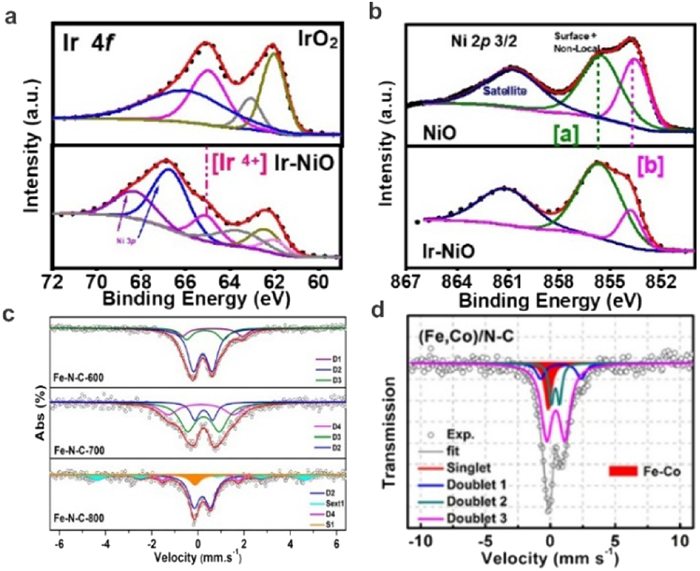
X-ray photoelectron spectroscopy (XPS) is widely applied for analyzing the chemical properties of the surface and the electron transfers between the catalysts and the substance. The XPS technique can be applied to collect the kinetic energy and electrons on the surface of materials in the range of 1 nm to 10 nm. In addition to determine the composition of elements in materials, the chemical and electronic states of the elements can be well studied by XPS. Zhao and co-workers employed a MOF based synthesis method to fabricate Ni single atom catalysts (NiSA/N—C) for efficient CO2 electro-reduction [37]. The XPS spectra show that the binding energy of the Ni 2p3/2 peak is higher than that reported for Ni0 (852.5 − 853.0 eV) and lower than that for Ni2+ (855.7 eV), indicating the presence of ionic Niδ+ (0 < δ < 2) in NiSA/N—C. Wang et al. studied the electronic structure of Ir and Ni in an single Ir atoms/NiO catalysts by XPS [40]. The XPS spectra for typical IrO2 show two sets of doublets centered at 62.1 and 65.0 eV, 63.1 and 66.0 eV; while for the IrSA-NiO, the Ir 4f spectrum can be deconvoluted into two sets of doublets centered at 62.3 and 65.1 eV, 61.9 and 63.8 eV, indicating the presence of Ir4+ and Ir3+ (Fig. 5a). For the Ni XPS spectra of IrSA-NiO, the main peaks of Ni 2p2/3 at 853.7 eV is much higher than that of bulk NiO, which indicates an increased valance state of Ni atoms on the surface (Fig. 5b). These results further proved the significant charge transfer between isolated atom and support.
XPS spectra are also used to study the electron structure of substrates. For the support of single atoms, N doped carbon-based substrates are one type of commonly used supports. The detailed carbon and doped N structure can be well distinguished according to the peak positions in XPS. For instance, Zhang et al. investigated the N-doping effect for the fabrication of Pt SACs on carbon nanotubes by preparing three different types of N-doped CNT as the substrates (denoted hereafter as CNT, NCNT and HNCNT). They found that the ratio of the C—C defect peak increased gradually with an increase amount in N-doping, indicating the creation of defects by N-doping in carbon support. By fitting the asymmetric N 1s spectra into six peaks, the different types (including pyridine-like nitrogen, pyrrole-like nitrogen, quaternary N) of N on carbon can be distinguished. It is found that the ratio of pyrrole-like nitrogen to pyridine-like nitrogen increased a lot with the doping of N in carbon nanotubes [41]. The presence of the high density of pyrrole-like nitrogen makes great contributions to the well dispersed Pt SAs and effectively enhances HER activity.
In the study of single-atom materials, work function measurement is crucial for understanding the electronic properties and behavior of materials. By measuring the work function of a single-atom material surface, the distribution and energy of surface electronic states can be revealed. The modification or doping of SACs can significantly change their electronic structure and properties. By measuring the change in work function before and after modification or doping, we can gain a deeper understanding of the impact of these processes on the electronic structure of materials.
The Mössbauer spectrum is a γ-ray absorption spectrum, which is collected when the energy of the incident γ photon is equal to the energy level transition energy of some nuclei in the material [42]. The Mössbauer spectrum can directly reflect the corresponding relationship between the number of γ photons and energy measured after absorption. The valence state of the atom, the ionicity of the chemical bond, and the coordination number of SACs can be determined according to the analysis of Mössbauer spectrum, due to the involvement of the energy level structure of the nucleus and the chemical environment by Mössbauer effect.
Liu et al. investigated the electronic structure of three different types of Fe-N-C catalysts (Fe-N-C-600, Fe-N-C-700 and Fe-N-C-800) by Mössbauer spectra. The results indicate that the relative concentration of each FeNx species is critically dependent on the pyrolysis temperature (Fig. 5c) [43]. For both Fe-N-C-600 and Fe-N-C-700 samples, three doublets can be observed in the Mössbauer spectra. In the Fe-N-C-800 sample, in addition to two doublets, one singlet (S1) and one sextet (Sext1) corresponding to γ-Fe and FexC species appear in the spectrum. They found that the concentration of doublets for low-spin N−(FeⅢN4)−N firstly increases with the pyrolysis temperature, and then it completely disappears in Fe−N−C-800. This result suggests that the doublets for low-spin N−(FeⅢN4)−N structure is destroyed and then aggregation of Fe0 occurs as a consequence of pyrolysis at higher temperatures (800 ℃ or above). For the medium-spin N−(FeⅢN4), the concentration gets maximized in Fe−N−C-700, which is confirmed to be the most active sites for selective oxidation of the C−H bond according to the DFT simulations. Besides SACs, the formation of dual atomic sites can also be confirmed by Mössbauer spectra. Wang and co-workers investigate the coordination environment of Fe-Co dual sites by Mössbauer spectra (Fig. 5d) [44]. Similar to the Fe single atoms, the spectrum of Fe SAs/N—C can be fitted with three doublets: Square-planar FeⅡN4 coordination with Fe(Ⅱ) in a high-, low-, and intermediate-spin state, respectively. For the Fe-Co dual atomic sites, the three FeN4-centers (D1-D3) can be observed similar to those of the FeSA/N—C. More specifically, the occurrence of the minor amount of iron species (singlet component) in (Fe, Co)/N—C demonstrates presence of Fe-Co bonding.
Reducing the size of the noble metal nanoparticles to clusters or even single atoms is an effective route for increase the utilization efficiency of catalysts and decrease the catalyst cost. Presently, many noble metal single atoms have been synthesized based on pyrolysis process, atomic layer deposition (ALD), galvanic replacement reaction, photochemical reduction and so on, which have been widely applied in many catalytic reactions [27,45-51]. The combination of DFT and synchrotron radiation techniques has played a pivotal role in elucidating the bonding energies between atoms and their supports in SACs. When DFT calculations are combined with EXAFS analysis, the resulting synergy provides a powerful tool for understanding the bonding energies and structures of single-atom materials. The XANES and EXAFS data can provide more detailed information about the atomic structure. This, in turn, enables researchers to validate and refine their DFT models, leading to a more accurate understanding of the bonding mechanisms.
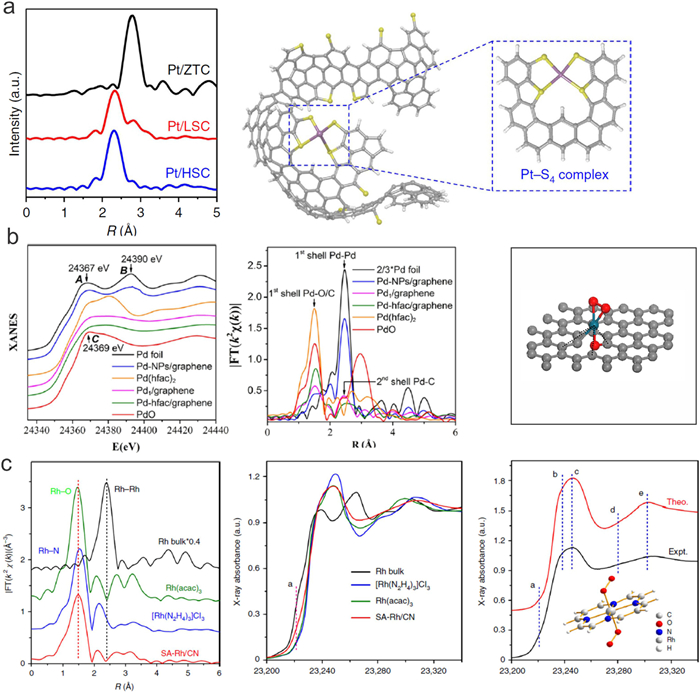
Wu et al. developed an iced-photochemical reduction method to load Pt atoms on various substrates, including mesoporous carbon, graphene, carbon nanotubes, titanium dioxide nanoparticles, and zinc oxide nanowires [26]. Pt single atoms exhibit positively charged state with the coordination of C/O species for Pt1/mesoporous carbon. When Pt is deposited on N-doped carbon substrates, Pt tends to anchor on the N sites [46,52]. Sun and co-workers deposited Pt single atoms on N-doped substrates by ALD methods. The EXAFS fitting results indicate that Pt single atoms have the CN of 2.2 and 1.2 for Pt-C and Pt-N bonding respectively, which agrees well with the proposed DFT model [53]. Besides Pt-C and Pt-N, Pt SACs are deposited on S-doped zeolite-templated carbon via a simple wet-impregnation method [49]. As shown in Fig. 6a, the abundant S-functionalities can bond with Pt atoms and a dominant EXAFS peak at 2.29 Å can be observed, which can be assigned to Pt–S coordination (CN=3.8). This result indicated that the coordination environment of Pt single atoms can be significantly tuned by doping other elements in pure carbon substrates. Ru-based nanomaterial is another noble metal, which has been applied in several catalytic reactions [54-58]. When Ru single atoms are deposited on mesoporous C3N4 via a simple wet impregnation method, the Ru sites bonded with N exhibit excellent activity and selectivity for both the hydrogenation and hydrodeoxygenation of vanillin [59]. In another study, Li and Wu used the strong coordination between Ru3+ and the UiO-66−NH2 structure to prepare Ru single atoms supported on nitrogen-doped porous carbon (Ru SAs/N−C) [60]. Due to the bonding energy and electron transfer between Ru single atoms and N—C support, the chemical state of Ru is detected close to 3+ according to the XANES results. The Ru−N/C has a coordination number of 3 and a mean bond length of 2.08 Å. When using Ru3(CO)12 as the precursor, the uniform Ru3 clusters are obtained on the substrates [61]. In this catalyst, the coordination number of Ru−N and Ru−Ru is 2.2 and 1.6, and the corresponding mean bond length is 1.99 Å and 2.53 Å. Pd have potential applications in electronics, dentistry, medicine, hydrogen purification, chemical applications, groundwater treatment, and jewelry [21,62-64]. Similar to Pt atoms, Pd atoms tend to bond with N when they are deposited on nitrogen-doped graphene by a freeze-drying-assisted method [65]. The FT-EXAFS fitting results clearly conclude the formation of Pd-N4 structure for Pd1/N-graphene, and XPS confirmed the presence of Pd2+ in Pd1/N-graphene. Pd single atoms can also be prepared on graphene by the ALD method [50]. The EXAFS spectra imply a high distorted structure around the nearest coordination of Pd atoms. The first shell peak for Pd1/graphene is contributed to the mixture of Pd−O (the oxygen atom bridging the Pd atom and the graphene support) and Pd−C coordination (the carbon atom from the graphene support). The fitting results show that bond lengths for Pd−C1, Pd−O1, and Pd−O2 are 2.00, 2.05, and 2.07 Å, respectively (Fig. 6b). Recently, Rh single atoms are developed as an anodic electrocatalyst in direct formic acid fuel cells [51]. In the XAS study (Fig. 6c), the Rh SACs exhibits a major peak at ~1.48 Å, which is shorter than the Rh–N peak at ~1.54 Å for the RhN6 octahedra in [Rh(N2H4)3]Cl3 and slightly larger than the Rh–O peak at ~1.45 Å for the RhO6 octahedra in Rh(acac)3. This result indicated that the Rh is coordinated with both N and O atoms in the sample of Rh SACs. In addition, the fitting analysis demonstrated that the coordination number of the N/O atoms in the first coordination sphere of Rh was estimated to be 6.3 at distance of 2.05 Å, indicating an octahedral configuration for the Rh–N/O bonding. DFT calculations suggest that the formate route is more favorable on SA-Rh/CN. According to calculations, the high barrier to produce CO, together with the relatively unfavorable binding with CO, contribute to its CO tolerance.
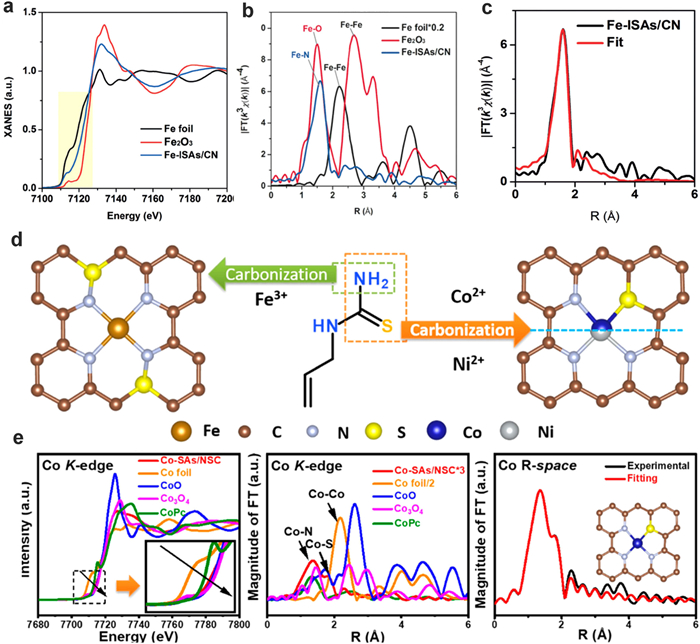
Recently, transition metal single atom materials have emerged as one of the most promising alternatives to precious-metal catalysts in several catalytic reactions [66-76]. Fe-based materials exhibited comparable catalytic activity with precious-metal catalysts in the oxygen reduction reaction (ORR) [67,68]. For example, Li and co-workers selected zeolitic imidazolate frameworks (ZIF-8) to separate and encapsulate the metal precursor Fe(acac)3. The isolated Fe single atoms can be anchored on nitrogen-doped porous carbon by pyrolyzing the ZIF precursors [36]. Due to the formation of Fe-C/N bond, the electrons transfer from Fe to substrates and Fe single atoms carry positive charges (Fig. 7a). The coordination number of Fe-N can be determined to be around 5 according to the fitting results of FT-EXAFS curve (Figs. 7b and c). The Fe-N coordination number can be tuned to 4 by anchoring Fe atoms on three-dimensional hierarchically porous carbon [69]. In addition to Fe-N structure, Fe-isolated single atoms were deposited on sulfur and nitrogen-codoped carbon (Fe-ISA/SNC) by pyrrole–thiophene copolymer pyrolysis strategy (Fig. 7d) [66,70]. In this case, although sulfur is not directly coordinated with Fe atoms, the doping of sulfur atoms has influenced the coordination environments of Fe and N atoms. The XAS and XPS results confirmed the formation of FeN4S2 active centers. The low electronegativity of S can enrich the charge on N atom, which facilitate the rate-limiting reductive release of OH*, and accelerate overall ORR process. The Fe electron environment is further tuned by coordinating with a nitrogen, phosphorus and sulfur co-doped hollow carbon polyhedron substrate [71]. According to the FT-EXAFS fitting results, the Fe atoms are boned with 4 N atoms, but not P nor S. Co SACs can also be synthesized by using MOFs methods, especially zeolitic-imidazole frameworks (ZIFs), as precursors through a thermal activation process [72]. For example, Wu and co-workers prepared Co single atoms anchored on ultrathin two-dimensional nitrogen doped carbon and core–shell structured Co–N–C catalysts [73,74]. The Co atoms are bonded with 4 N in a tetrahedral geometry on the substrates. Besides the deposition of Co atoms on the N-doped substrates, Co atoms are anchored on porous N, S-doped carbon (NSC) matrix to further tune the bonding energy of Co and the support [66]. On NSC matrix, the Co single atoms formed one Co-S bond and 3 Co-N bond. In the CoN3S1 (three Co−N bonds and one Co−S bond) structure, the Co-S bond length is 0.46 Å longer than the Co−N distance (Fig. 7e). When Co single atoms are deposited on N and P co-doped carbon support, only Co-N coordination can be observed according to the FT-EXAFS curve [72]. The Co-P shell at near 2.0 Å was not observed, confirming that there was no Co-P contribution in the Co1/P-NC catalyst. Besides, Wu and co-workers prepared Mn single atoms on partially graphitic carbon [75]. The Mn atoms represent a combination of the Mn–N and Mn–C scattering paths in the FT-EXAFS spectra with the coordination number of 4. DFT calculations confirm that the MnN4C12 site has a favorable binding energy with O2, OOH and H2O during the ORR as well as a surmountable energy barrier to break O–O bonds for complete 4e− oxygen reduction, which results in the superior performance of Mn-SAC in the PEMFCs application. Zn single atoms are also fabricated by pyrolyzing carbon black, urea, and zinc acetate [76]. The electron transfer between Zn and N in the Zn-N4 sites can effectively enhance the activity for CO2 reduction and ORR.
Besides carbon-based substrates, transition metal oxides are also widely applied as a typical substrate for the deposition of single atoms. As single atoms are usually bonded with the O sites of the metal oxide, the single atoms exhibit partially positively charged electronic state [77-79]. For instance, Prof. Zhang et al. uniformly dispersed Pt single atoms on a FeOx support with high surface area by a co-precipitation method (Fig. 8a) [77]. The whiteline intensities of Pt in the XANES spectra suggests that the Pt single atoms carry positive charges in the degree between Pt and PtO2 (Figs. 8b and c). The high density of d orbitals of the single Pt atoms and strong interaction with FeOx are account for the excellent catalytic activity for both CO oxidation and preferential oxidation (PROX) reactions. When Ir is deposited onto FeOx substrates, the Ir species also become more positively charged due to the interaction and electron transfer occurs between Ir single atoms and FeOx substrate [80]. The positively charged Ir single atoms are the most active sites for water gas shift. TiO2 is another typical metal oxide for the deposition of metal single atoms, such as Pt, Ir and Pd. Wang and Li report the positively charged Pt single atomic sites on anatase TiO2 catalyst with remarkable activity, optimal selectivity and excellent reusability toward anti-Markovnikov alkene hydrosilylation due to the atomic dispersion of active species and unique partially positive charge Pt electronic structure (Figs. 8d-f) [81]. Lei et al. stabilized Pd single atoms on TiO2 thin film and found that as-prepared Pd atoms are coordinated with 4 chlorine atoms [82]. Wang and Li deposited Ir single atoms on TiO2 and mesoporous graphitic C3N4 [83]. The Ir atoms only bond with N/O/C in the Ir1/C3N4. For the Ir1/TiO2, in addition to the Ir-O bond, Ir−Ti bond also exists according to the EXAFS analysis. Recently, The loading amount of Ir was increased to ~18 wt% on NiO substrates [40]. The coordination distance of Ir-Ni is shorter than Ir-Ir and existents as the Ir-O-Ni in the Ir-NiO structure. The Ni-O is shortened in Ir-NiO compared with pure NiO substrate, which might cause strong bonding interaction and between Ni and O atoms. In addition to FeOx, TiO2 and NiO, ZnO nanowires were employed as the support materials to deposit Rh single atoms [84]. In this case, the electrons transfer from partially reduced ZnO to Rh, and result the Rh species exist in an almost metallic state, which is concluded by XAS and XPS results. The obtained Rh SACs on ZnO nanowires exhibited a turnover number (TON) of about 40,000 and 99% selectivity during the hydroformylation process.

Besides metal oxides, the layered double hydroxides (LDH) were another type of metal compounds for deposition of single atoms [85]. For example, Sun and co-workers fabricated a strong electronic coupling by anchoring Ru single atoms on the surface of CoFe LDH [86]. In this case, the EXAFS spectra prove the formation of Ru–O–M structure. The model-based EXAFS fitting further confirms that each Ru atom is coordinated with 4 oxygen atoms, in which 3 Ru–O were bonded nearby Co or Fe metal. Zhang et al. fabricated Au single atoms on NiFe LDH (Au/NiFe LDH). In the XANES spectra, the white line peak of Au/NiFe LDH is sharp; while the Au foil exhibit almost no white line peak due to the completely filled 5d state of Au (Figs. 8g and h) [87]. This result suggests that Au/NiFe LDH have a positively charged Au atomic local structural environment compared to that of metallic Au, which results in the significantly enhanced activity of Au/NiFe LDH towards the OER (Fig. 8i). The strong metal-support interaction can also be created on substrates of metal carbides (MoC), metal sulfides (MoS2) [45,88,89]. For example, Ma et al. report that Pt atomically dispersed on α-molybdenum carbide (α-MoC) by using temperature-programmed carburization (TPC) method. The interaction between metal and support substantially changes the electronic structure of Pt and α-MoC. XPS results show that the binding energy of Pt 4f7/2 is about 0.6 eV higher than that of metallic Pt. DFT results also show that the electron density at Pt sites reduces greatly compared with that over metallic Pt, which benefit the aqueous-phase reforming of methanol reaction. Recently, MXenes, a large family of emerging two-dimensional (2D) materials, have been considered as a new type of substrate for stabilizing single atoms. Wang and co-workers used double transition metal MXene nanosheets (Mo2TiC2Tx) for deposition of Pt atoms, in which the Mo vacancies can act as the anchoring sites for single Pt atoms [90]. The Pt atoms exhibit positively charged properties, which can increase the hydrogen adsorption, leading to faster formation and release of molecular hydrogen.
The deposition of single atoms on metal substrates can bring the catalysts with specific physical and chemical properties due to the formation of bimetallic bonding structures [84,91-93]. Recently, Pt single atoms are deposited on Cu and Pd substrates and their electronic structure are tuned different from that on metal oxides or carbon substrates. The distinct coordination environments of SAA can be observed from FT-EXAFS spectrum. Wei and Wu found that the Pt-Cu SAA samples have one prominent peak in the region of ~2.2–2.3 Å attributed to the formation of Pt-Cu bond, which is located between PtO2 standard sample and Pt foil. During the catalytic glycerol hydrogenolysis to 1,2-propanediol process, the interface sites of PtCu–SAA serve as intrinsic active sites, in which the single Pt atom facilitates the breakage of central C–H bond and the Cu atom promotes the dissociation adsorption of terminal C–O bond [94]. The similar electronic structure changes of Pt single atoms have also been studied by Sykes and co-workers [95]. The Pt atoms on Cu geometries are found to exhibit high activity and selectivity for butadiene hydrogenation to butane under mild conditions. When Pt is deposited onto shape-controlled Pd surfaces, the electrons transfer from Pt to Pd, resulting in the formation of a peak located at 2.2 Å in the R-space spectrum of EXAFS of Pt/Pd SAA catalysts (Figs. 9a and b) [96]. This tuned structure of Pt SAA facilitates the activity by reducing the adsorption energy of OH* during the ORR process.
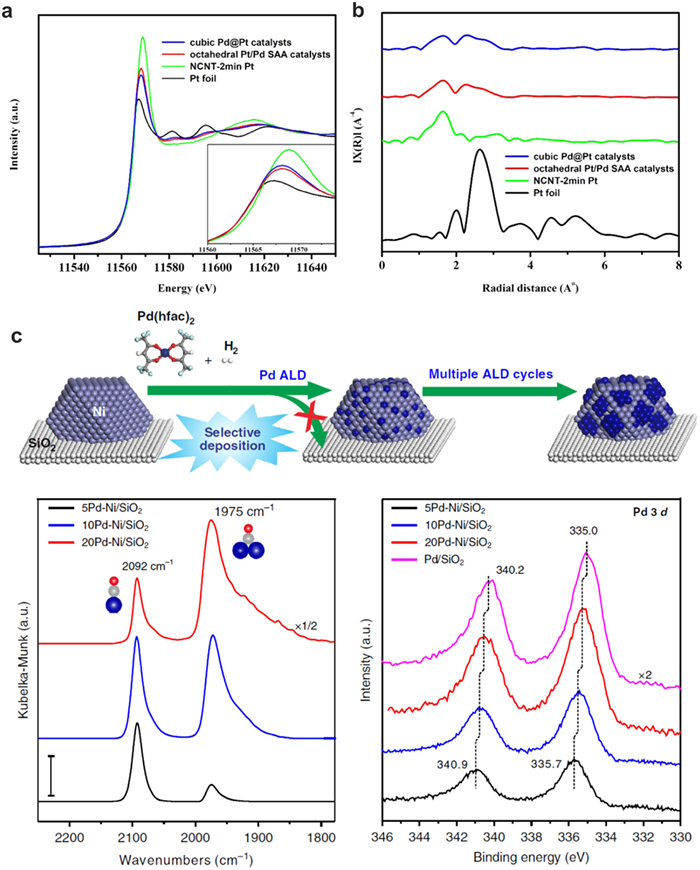
Besides Pt atoms, Pd atoms were also deposited onto metal substrates for specific catalytic reactions. For example, Lu and co-workers synthesized atomically dispersed Pd on Ni nanoparticles to break the strong metal-selectivity relations in benzonitrile hydrogenation [97]. The Pd-Ni coordination information is carefully studied by the EXAFS spectrum of Pd1nullNi/SiO2. The dominant peak at 2.12 Å and corresponding fitting results reveal that the Pd-Ni coordination number is 5.5, while the Pd-Pd coordination number is only 1.2, suggesting the formation of a core-shell structure like quasi Pd1Ni SAA structure (Fig. 9c). The bonding energy between single atoms and supports in single-atom catalysis is a key factor in determining the stability, activity, and selectivity of the catalyst. The nature of the support material can significantly influence the electronic structure of the single atom, leading to unique catalytic properties. Strong bonding between the single atom and the support can enhance the durability of the catalyst, while weaker bonding may allow for dynamic changes in the catalytic site during the reaction. Understanding and controlling the bonding energy in single-atom catalysis is crucial for designing efficient and stable catalysts for a wide range of chemical processes.
Besides the SACs, sub-nano metal clusters with specific atoms received more and more attentions due to their unique and unexpected properties [98-102]. The synergistic effect of the homonuclear atoms may modulate charge transfer between two single atoms and result in a high catalytic activity by optimizing the adsorption energy with reactants. For example, Wang and co-workers reported a "precursor-preselected" wet-chemistry strategy to synthesize highly dispersed Fe2 clusters on mesoporous carbon nitride (mpg-C3N4), which exhibits superior catalytic performance for the epoxidation of trans-stilbene to trans-stilbene oxide [103]. They used XAFS spectroscopy to probe detailed structure information such as the coordination environment of the Fe2 structure. Interestingly, the two Fe atoms exhibited different positive charges of 1.32 and 1.00, respectively, according to the Bader charge analysis. In the FT-EXAFS curve of Fe2/mpg-C3N4 sample, a strong peak can be observed at ca. 1.53 Å, suggesting that the sample is mainly comprised of the Fe–N/O coordination path. In addition to the Fe–N/O peak, a secondary peak at high R value (ca. 2.27 Å) reveals the formation of Fe–Fe path for the surrounding coordination of metal centers. The FT-EXAFS fitting results indicated that the average coordination numbers of Fe–N/O and Fe–Fe are 3.8 and 1.2, respectively, further confirming the structure of Fe2. Xing and co-workers synthesized the binuclear Co2Nx site by controlling the catalyst structure at molecular level [32]. Atomic resolution STEM images show the successful fabrication of two conjoint Co ions with a Co-Co distance of 2.1–2.2 Å. The FT-EXAFS curve shows two main peaks at 1.6 and 2.1 Å, which can be assigned to Co-N and Co-Co coordination, respectively. The WT-EXAFS signals indicated that the Co-Co distance in the Co2N5 is shorter than that in the Co nanoparticles. The Co2N5 sites can pre-absorb OH as a modifying ligand to promote the reaction of *OH → H2O, thus exhibited > 12 times higher activity than the conventional CoN4 active site.
In addition to the synthesis method by pyrolyzing MOF-based catalysts, the ALD technique provides the possibility to achieve atomically precise ultrafine metal dimers, even bimetallic sites. For example, Lu and co-workers prepared Pt2 dimers on graphene by a bottom–up approach using ALD method [104]. They found that the Pt2 dimers are likely in the oxidized form of Pt2Ox with O atoms alternating between the terminal and bridge positions. In the EXAFS analysis, no obvious Pt-Pt bond was observed. The simulation result indicated that Pt–Pt bond distance in the Pt2O6 chain is 3.03 Å, consistent with the experimental results very well. It was found that the unoccupied 5d states of the top Pt atom in Pt2/graphene-R locates at a considerably higher energy position of 0.87 eV above Fermi level than that of the Pt atom in Pt1/graphene-R (0.40 eV). Compared to Pt single atom, the higher energy position of the unoccupied state of the Pt 5d orbital of the top Pt in the Pt2 dimer might play an important role in weakening the adsorption of both ammonia borane and H2 molecules, thus facilitating the hydrolytic dehydrogenation of ammonia borane activity.
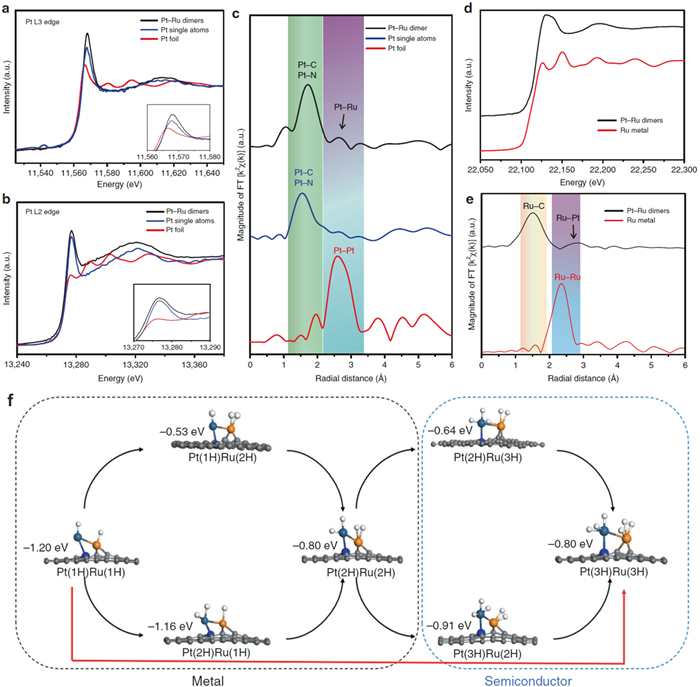
Recently, heteronuclear dual-metal sites are found to exhibit greatly enhanced catalytic activities, which is due to the synergistic effect and electron transfer between the two neighboring metal sites. Sun and co-workers developed the novel ALD route to generate Pt-Ru heteronuclear dimers on nitrogen-doped carbon nanotubes (NCNTs). The electronic structure of Pt and Ru atoms is carefully studied by the XAS and DFT (Fig. 10) [53]. For the FT-EXAFS of Pt L3 edge, an obvious peak at 1.6 Å is attributed to Pt-N/C bond. In addition, a relative weak feature resolved at around 2.6 Å is assigned to the Pt1-Ru1 dimer structure (Fig. 10c). The EXAFS R space curve fitting result indicated the coordination number of 1 for Pt1-Ru1 dimer. Furthermore, from the Ru R space fitting results, the Ru atoms have the CN of 0.7 for Ru-Pt, both suggesting the successful fabrication of Pt1-Ru1 dimer structure (Fig. 10e). The calculated hydrogen adsorption Gibbs free energies (ΔGH) of the Pt1-Ru1 dimer were performed to examine the catalytic activity of the HER for different number of H atoms. The Pt1-Ru1 dimer generates the synergy effect by modulating the electronic structure which evolves from metallic to semiconducting during the hydrogen adsorption, resulting in an increase in the unoccupied orbitals and a decrease of the bonding between the Ru and H. The Ru atom exhibits the low ΔGH of 0.01 eV through the pathway of Pt(3H)Ru(3H) → Pt(3H)Ru(2H), which clearly indicates that the hydrogen atom becomes easy to detach from Ru when the maximum coverage of 6H is reached (Fig. 10f). Thanks to the well-tuned electronic structure of Pt and Ru atoms, the as-prepared Pt-Ru dimers show significantly enhanced mass activity and stability compared with commercial Pt/C catalysts for the HER.
By pyrolyzing chitosan and zinc chloride and cobalt acetate, Zhao and Sun fabricated Zn-Co atomic pairs anchored on N-doped carbon support [105]. According to the EXAFS fitting results, the Co-N coordination number is 3.5 and the coordination number of Co-M is 0.5 ± 0.1. For the Zn K-edge EXAFS, the Zn-N coordination number is 3.5 in the first shell, together with a weak second shell due to Zn-M (CNZn−M = 0.5 ± 0.1), confirming the existence of Zn1nullCo1 atomic pair bonding in Zn/CoN—C. The DFT results indicate that the *OH reduction on ZnCoN6 is an exothermic process with high stability, which enables the *OH to act as a modifying ligand forming a ZnCoN6-(OH) structure. Besides Zn-Co bimetallic dual atomic sites, Fe-Co dual sites embedded on N-doped porous carbon ((Fe, Co)/N—C) was reported exhibited good ORR performance in acidic and alkaline electrolyte [44,106]. The electronic structure and coordination environment are studied by XAFS). A strong FT-EXAFS peak of Fe K-edge located at 1.51 Å is attributed to the Fe-N coordination path. A secondary peak at ca. 2.25 Å demonstrated the metal-metal path which should be accounted for the surrounding coordination of metal centers in the first shell. According to fitting results, coordination numbers of Fe-N and Fe-Co are calculated to be 3 and 1, respectively. The DFT fabricated model with the Fe-Co bond agrees well with the experimental spectra, further confirming the formation of Fe-Co dual atomic sites. Zhao et al. synthesized isolated diatomic Ni-Fe sites anchored on nitrogenated carbon by a dual-solvent route [107]. Similar to other diatomic pairs, the EXAFS spectra show the dominant Ni-N coordination and a metal-metal path for the formation of Ni-Fe coordination. They also found that the rate-determining step for Ni/Fe-N-C during carbon dioxide electro-reduction (CRR) is the first electron transfer, which generates surface adsorbed *COOH intermediate. In some other cases, the synergistic effects may come from two individual atomic sites. For instance, Zhu et al. found the Zn-Co monomers exhibited extremely high CO2 electroreduction selectivity to CO [108]. The EXAFS spectra and corresponding fitting result indicated the co-existence of Co-N4 and Zn-N4 structure, but not Co-Zn bond on the supports. The DFT calculation demonstrated that the *COOH adsorbs on Co sites and converted to *CO, then *CO transfer to Zn sites and desorb from Zn during the CO2 reduction process.
In summary, the bonding energy between isolated atoms can promote the progress of the catalytic reaction. In addition, two heteronuclear atoms play different roles in catalytic reactions. The interaction between two single atoms can adjust the electronic structure of the active center, thereby affecting the catalytic performance. Moreover, the relative spatial positions of the two single atoms also affect the catalytic performance. Appropriate geometric arrangement can provide more active sites, promoting the adsorption of reactants and the desorption of products.
Bonding energy between single atoms and reactants is a critical factor in understanding and predicting the reactivity and stability of single-atom catalysts. The bonding energy between a single atom and a reactant is dependent on the properties of the reactant itself, such as its electronegativity, electron configuration, and functional groups present. It determines the ease with which the reactant can adsorb onto the catalyst surface, and influences the selectivity of the catalytic reaction by determining which reactants or intermediate species are preferentially adsorbed and stabilized on the catalyst surface. Thermocatalytic reactions are important chemical and chemical processes, which are of great significance in both fundamental research and industrial applications. In single-atom catalysis area, the research focus of thermocatalytic reaction is to explore the catalytic reaction mechanism and insight the enhancement of catalyst activity.
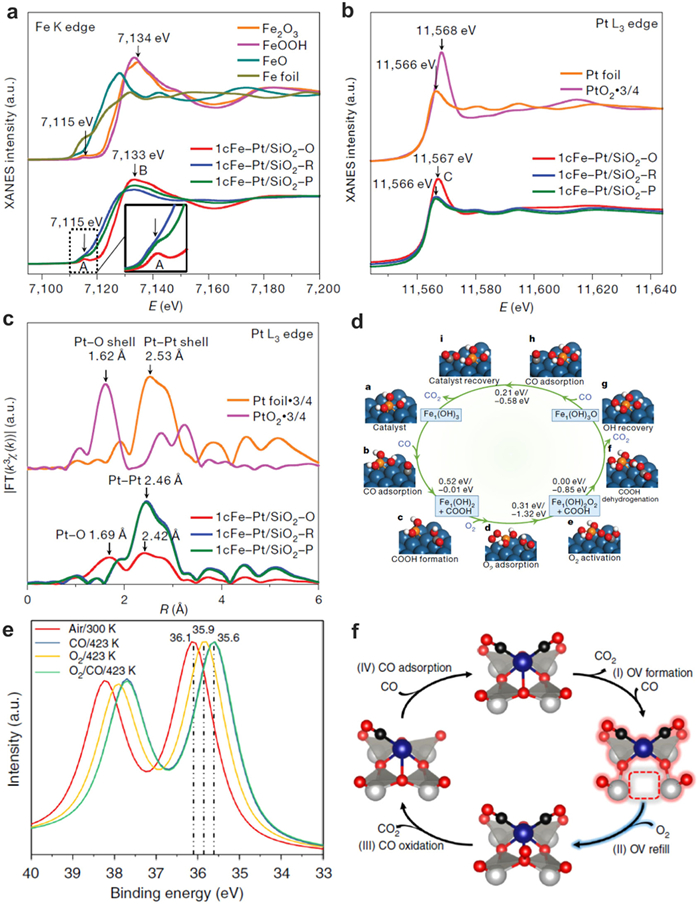
During the reaction progress, bonding energy of SACs and reactants can be detected by in-situ technique. Zeng and co-workers reported the synergetic interaction between neighboring Pt monomers. The Pt/MoS2 catalysts are prepared via replacing Mo atoms in MoS2 nanosheets with Pt atoms [88]. The 7.5 wt% Pt/MoS2 with the 65.5% neighboring Pt monomers exhibited distinct catalytic selectivity for CO2 hydrogenation compared with isolated monomers on 0.2 wt% Pt/MoS2. The DRIFT and XPS measurements indicated that two intermediate species, COOH* and CH2OH*, were observed for both 0.2 wt% Pt/MoS2 and 7.5 wt% Pt/MoS2. In addition, the main intermediates for 0.2 wt% Pt/MoS2 and 7.5 wt% Pt/MoS2 were CH2OH* and COOH*, respectively. The intermediates and reaction channels for CO2 hydrogenation over Pt1/MoS2 and Pt2/MoS2 were further simulated by DFT. On the Pt2/MoS2, the energy barriers for the transformation of COOH* into HCOOH is lower than C(OH)2*. Thus, HCOOH is more favorable and it will transfer to CH(OH)2*, CHOH*, CH2OH*, CH3OH in the subsequent steps. Prof. Lu and co-workers fabricated isolated Fe(OH)x on Pt/SiO2, which enables complete and 100% selective CO removal through the preferential oxidation of CO in hydrogen (PROX) reaction over the broad temperature range of 198–380 K [109]. The in-situ XAFS technique are used for detecting the change of electronic structure catalysts during PROX reaction (Figs. 11a-c) [110]. A home-made quartz reaction cell with Kapton foil was used to hold the catalyst pellet and this cell can be heated to 773 K with external heating. The XAFS spectra recorded according to the various reaction temperature and different atmospheres treatment can clearly analyze the electron transfers between Fe and reactant. The detailed reaction mechanism is simulated on the Fe1(OH)3@Pt(100) model catalyst (Fig. 11d). Firstly, CO adsorb on a bridge site of the Pt(100) surface and diffuse to a Pt atop site. Then CO will react with the elongated OH in Fe1(OH)3 to form COOH* and Fe1(OH)2. After Fe1(OH)2 has formed, one O atom of molecular O2 firmly bonds to the Fe atom while the other O atom bridge-bonds to two surface Pt atoms, and then this chemisorbed O2 can readily dissociate to yield Fe1(OH)2O2. The Fe1(OH)2O2 will react with COOH* via proton transfer to form Fe1(OH)3O@Pt(100). Subsequently, another CO adsorbs on the Pt surface and attacks the remaining O atom, producing the second CO2. This step regenerates the initial Fe1(OH)3@Pt(100) catalyst structure and closes the catalytic cycle, with the calculations indicating that association of CO with Fe1(OH)3@Pt(100) to form COOH* is the rate-determining step.
Besides, the bonding interaction and reaction mechanisms are also investigated on the single atoms in CO oxidation reactions [110]. Yan and co-workers investigated the structure and bond energy of atomically dispersed Rh on phosphotungstic acid with CO by a suite of operando/in situ spectroscopic experiments [10]. During the reaction, the catalysts are treated in CO flow with temperature stepwise-increased from 300 K to 473 K. The structure and reaction intermediates are investigated by XAS, XPS and diffuse reflectance infrared Fourier transform (DRIFT) spectroscopy. In the in-situ DRIFT spectroscopy, they found two CO frequencies at 2110 and 2039 cm−1, assignable to Rh(CO)23+ species. With the increase of temperature, the Rh(CO)23+ species transfers to Rh(CO)2+ species (Fig. 11e). When the reduced catalyst is treated under a dilute oxygen atmosphere, a partial oxidation is observed at around 323–373 K. Before complete re-oxidation, there is a subsequent removal of CO from Rh atoms (Fig. 11f). During the whole reaction, the oxygen vacancies on the single atom structure played an important role for the enhanced mechanism of CO oxidation.
In addition to the relationship between single atoms and reactants in thermo catalytic reactions, the reaction mechanism in electrochemical reactions is also investigated by the in-situ characterization and simulation [8,9,11,12,31,33,111-114]. By analysis the electronic structure of SACs at various reaction potentials, the catalytic reaction pathway can be figure out for different reactions, such as ORR, CRR, HER and oxygen evolution reaction (OER).
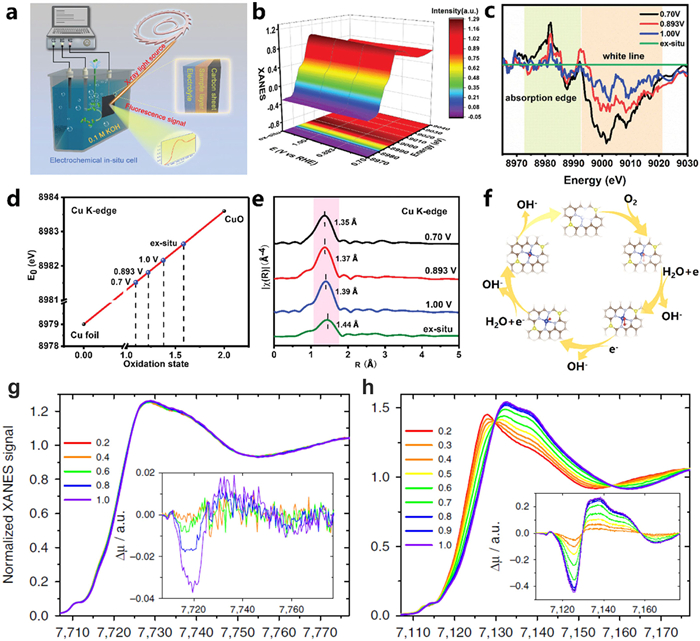
The improvement of catalytic activity in the ORR is significant to the development of proton exchange membrane fuel cells, metal-air batteries and other oxygen involving energy conversion devices. The non-noble metal nanomaterials have been widely investigated as effective electrocatalysts for the ORR. For example, Li and co-workers proposed an atomic interface strategy to fabricate single atom Cu catalyst on a nitrogen modified carbon support (Fig. 12a) [111]. The FT-EXAFS and corresponding fitting analysis of Cu-SA/NC suggest that the Cu species in Cu-SA/NC exist in the atomic coordination of Cu1–N4–C8S2. Furthermore, the interaction between Cu atomic sites and O2 is studied by detecting the structural evolution of Cu under electrocatalytic conditions via potential-dependent in situ XAFS measurements (Figs. 12b and c). The results show that the absorption edge of Cu was gradually shifted to lower energy, and the white line peak intensity reduced with the applied potential reduced from 1.0 V to 0.7 V. The average chemical valence of Cu decreased from 1.59 to 1.09 under the working conditions, which implied that the relative low oxidation valence of Cu (+1) species work as the catalytic active sites during the ORR process (Fig. 12d). The real-time FT-EXAFS at the Cu K-edge indicated that the Cu–N4–C8S2 moiety were respectively adsorbed with OOH*, O* and OH* intermediates under 1.0, 0.893 and 0.7 V (Fig. 12e). DFT calculation results indicated that for the Cu–N4–C8S2 interface in which S atoms are located at the opposite sites of the Cu central atom in the same hexatomic rings. All the elementary steps, including the formation of OOH* and O* are exothermic dramatically, which are more favorable for the ORR process compared with those on Cu–N4–C10 (Fig. 12f). The formation of bond-shrinking low-valence HOO–Cu1–N4–C8S2, O–Cu1–N4–C8S2 and HO–Cu1–N4–C8S2 sites under working conditions is the key role for the high ORR activity of Cu-SA/SNC. Zitolo et al. synthesized atomically dispersed Co atoms by a pyrolytic method and identified the structure and electronic state of three porphyrinic moieties during ORR, CoN4C12, CoN3C10, porp and CoN2C5 by the operando X-ray absorption [114]. When compared with Fe-N-C catalysts, the Co-based active moieties are unmodified under the external potential conditions from 0.0 to 1.0 V vs. RHE, while Fe-based moieties experience structural and electronicstate changes (Figs. 12g and h). DFT calculations showed that the single atomic Co atoms might bind oxygen intermediates too weakly relative to Fe moieties. Therefore, modifications of the carbon matrix with nitrogen or other hetero elements to increase the binding energy with O2 at Co centers should favor in enhancement of their ORR activity.
The OER, which represents a key half-reaction in water electrolysis, has enormous impact on the overall energy efficiency. However, OER still suffers with sluggish kinetics. Till now, the most efficient OER catalysts are still noble metal or Ru and Ir-based oxides that are high in cost and scarce in natural resources. Due to the advantages of 100% metal atom utilization efficiency, the application of SACs is considered as a frontier strategy for design the advanced OER electrocatalyst to overcome the challenges of noble metal-based catalysts [40]. Sun and co-workers prepared a stable single atomic Ru catalyst anchoring on the surface of CoFe-LDH, which possesses a strong electronic coupling between Ru SAs and CoFe-LDH support. The Ru1—CoFe-LDH catalyst exhibits outstanding activity with overpotential 198 mV at the current density of 10 mA/cm2 and a small Tafel slope of 39 mV/dec for the OER. To further investigate the role of monatomic Ru atoms and CoFe-LDHs regarding OER activity and stability, the powerful in-situ and operando XAS was carried out to probe the structural and oxidation state changes of active centers under the electrochemical conditions. The XANES found that Ru K-edge shifted to higher energy when the applied potential increased to 1.6 V vs. RHE, suggesting that the electron transfer of Ru atoms occurs during the OER. With decrease of the applied potential to initial stage, the XANES of Ru K-edge could shift back to the low energy position. However, the oxidation state of central Ru sites will not increase to higher than +4, which means that the single atomic Ru in Ru1—CoFe-LDH catalyst will not transform into an unstable phase of Ru. For Co and Fe, the Co and Fe in Ru1—CoFe-LDH catalyst have higher oxidization state than 2+ and 3+ at the open-circuit voltage (OCV). The oxidation state further increased during the OER process. DFT results indicated that for CoFe-LDH and R Ru1—CoFe-LDH structures, the OER rate-determining step was the formation of *OOH group from *O group. The Ru atom sites on the surface of CoFe-LDHs showed a lower Gibbs free energy (1.52 eV) of the rate determining step than that of the Fe atom sites on the edge of CoFe-LDHs (1.94 eV), revealing a more favorable OER kinetics on the Ru1—CoFe-LDH structures.
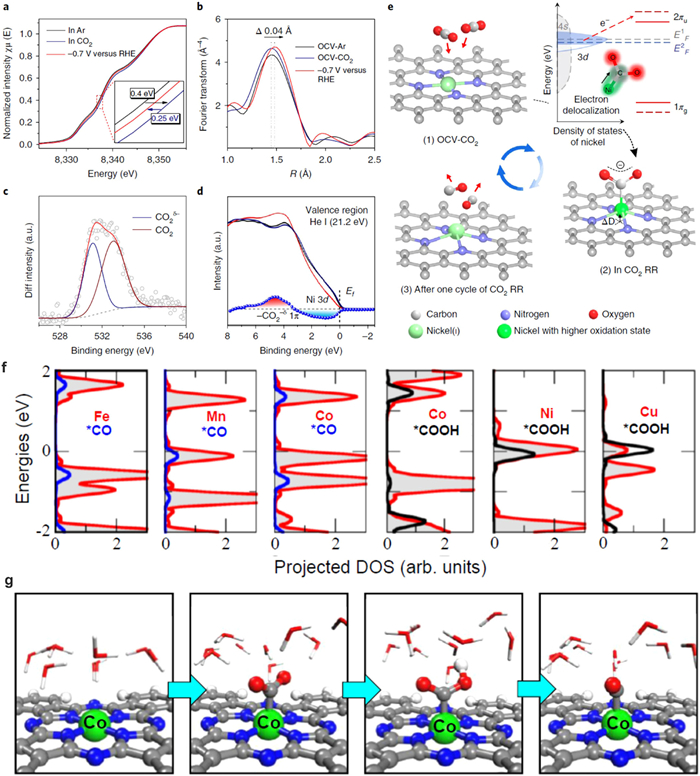
Recently, single atomic transition metals (Fe, Co and Ni) were found exhibited superior CO selectivity during electrochemical catalytic CO2 reduction [9,31,33,113]. Liu and co-workers fabricated Ni SACs (A-Ni-NG) by pyrolyzing a mixture of amino acid, melamine and nickel acetate [33]. The obtained isolated Ni atoms exhibit 97% CO Faradaic efficiency at an overpotential of 0.61 V. The electronic structure and coordination structure of Ni SACs is studied by Ni K-edge of XANES spectra at three different environments: in Ar-saturated KHCO3 solution under open-circuit voltage bias, in CO2-saturated KHCO3 solution under open-circuit voltage bias and in CO2-saturated KHCO3 solution biased at − 0.7 V vs. RHE (Fig. 13). It is found that the Ni oxidation state increase when CO2 is introduced into the KHCO3 solution under open-circuit voltage (Fig. 13a). This phenomenon is due to the delocalization of the unpaired electron in the 3dx2−y2 orbital and spontaneous charge transfer from Ni(ⅰ) to the carbon 2p orbital in CO2 to form a CO2δ− species. When the overpotential was set to −0.7 V, the Ni K-edge of A-Ni-NG shifted back to lower energy, which indicates the CO desorption and the recovery of low-oxidation-state Ni. In addition, the intensity of the EXAFS spectrum increased slightly and shifted (~0.04 Å) to longer coordination lengths at − 0.7 V, which suggests the expansion of Ni–N bond due to distortion of Ni atoms out of the graphene plane (Fig. 13b). In addition, DFT calculations also indicated the increase in the density of states around 3–6 eV below the Fermi level results from the 1π orbital of the bent CO2 molecules adsorbed on Ni(ⅰ) and the delocalization of the Ni 3d orbital. Based on the above analyses, the active sites can be concluded to be the Ni active site and the whole structural evolution process can be simulated in the electrochemical CO2 reduction (Fig. 13e). Prof. Deng investigated the CO2 reduction performance of a series of single atomic active metal centers (Mn, Fe, Co, Ni, and Cu) [113]. Among the different elementals, CoPc exhibited the highest Faradaic efficiency of 99% for CO2 reduction to CO with an applied potential of −0.8 V (vs. RHE). The electronic structure is studied during CO2 reduction at different potentials (Fig. 13f). The results show no significant changes in both XANES profiles and EXAFS spectra at the Co K edge of the CoPc catalyst when conducting −0.6 V for CO2 reduction, indicating that the valence state and the coordination structure of Co2+ were remained at the reduced potential condition (Fig. 13g).
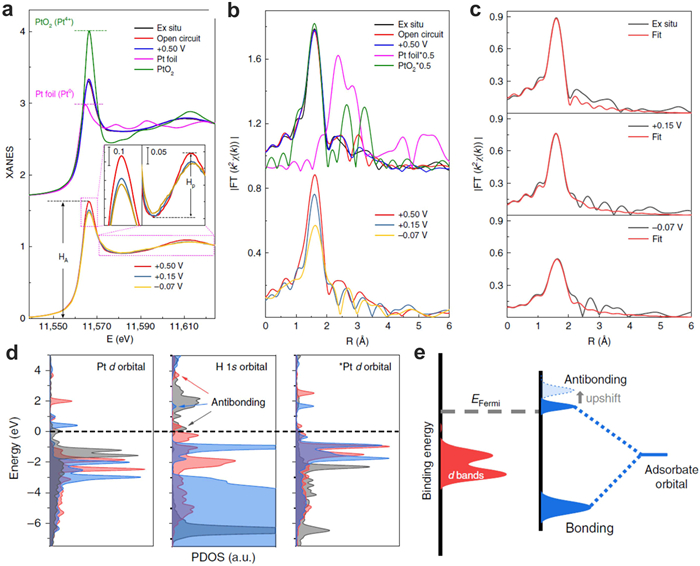
The electrochemical HER is an emerging route for curbing CO2 emissions while producing clean hydrogen. Pt is considered as the most effective electrocatalysts for the HER. However, its low abundance, limited supplies, and rising prices have motivated researchers to look for alternate catalysts with a high performance in the HER. Recently, it is found that Pt SACs exhibited extremely high mass activity compared with commercial Pt/C catalysts. For example, Sun and co-workers reported a practical synthesis method to fabricate Pt single atoms on graphene nanosheets by the ALD method. The as-prepared Pt single atoms exhibited 37 times enhanced electrochemical activities compared with commercial Pt/C catalysts [14]. Yao and co-workers investigated the near-free Pt single-atom dynamics during electrochemical HER by using Pt single atoms on UiO-66-NH2 [8]. In-situ XAS was carried out to identify the bonding energy of Pt SACs and reactants during HER process. When the applied potentials change from +0.15 V and to −0.07 V, the overall white-line intensity of Pt L3 edge decreases, indicating a higher 5d occupancy of Pt. This result implies the less charge transfer from the single-atom Pt to the nearby C/N atoms at +0.15 V and −0.07 V. In addition, the oxidation state of Pt decreases from 1.89 to 1.17 and 1.12 for the catalyst at the potentials from +0.5 V to +0.15 V and −0.07 V, respectively, suggesting that the Pt site is reduced close-to-zero valence toward the metallic state. Operando EXAFS results show that the coordination number of Pt-C/N decreased with the applied potential changed from +0.15 V to −0.07 V, which means that Pt atoms are released from support (Figs. 14a-c). Due to the small scattering amplitude of H, the detailed H adsorption process cannot be detected by the Pt–H coordination. Furthermore, the near-free Pt state during electro-reduction reaction can be observed in wide-pH environments, which played a key role for boosting the HER activity (Figs. 14d and e). Based on the above discussion, the bonding energy between single atoms and reactants can effectively determine the catalytic activity and efficiency of single-atom catalysts. This energy reflects the strength of the interaction between the catalyst’s active site, typically a single atom, and the reactant molecules. Understanding and tuning the bonding energy between single atoms and reactants is essential for designing highly active and selective single-atom catalysts for various chemical transformations.
The catalytic performance of single atom catalysts is highly dependent on the electronic structure of isolated atomic sites. The XAS, XPS and Mössbauer spectroscopy can determine the electronic structure and coordination environments of the single atoms. In this review, we demonstrated the detailed electronic structure of single atom catalysts and discussed bonding energy between the single atom-support, single atom-single atom, and single atom-reactants in single atom catalysis. The interaction between single atoms and support provides the strong force to anchor single atom sites and the bonding energy of single atoms and support can be examined by the XAS and DFT techniques. The formation of dimer structures plays a significant role in tuning the coordination environment of central metal atoms, which proves that the synergistic effect between neighboring bimetallic atoms contribute to improve the activity to a new level. The in-situ techniques provide deep insight information for further understanding the bonding energies between single atom sites and reactants/intermediates during the reaction condition. In the future, more research work should be directed to the following aspects:
(1) The studies about the electronic structure of metal single atoms, dimers, trimers, clusters and particles. With the development of synthesis methods, the atoms might be accurately controlled in the number from one atom to multi-atoms. It will be important to explore the electronic structures and their difference from single atoms, dimers to clusters and particles. In addition, the multi-atom catalysts can be heteronuclear atoms. With the formation of A-B atomic bonding, the electron transfer occurs and the electronic structure of metal atoms can be tuned, resulting in the excellent catalytic activity of SACs. The bonding environments of different atoms can be determined by XANES and EXAFS, which have provided the accurate coordinated atoms and coordination numbers of A-A dimers/trimers and A-B dimers.
(2) In-situ studies of the electronic structures and coordination during the deactivation process of single atom catalysts. Due to the extremely high surface energy, the atoms on support might aggregate and their interior structures will change during the catalytic reactions. The in-situ techniques have been used for identifying the electronic structure change during one cycle of reactions. In the future, it is of great importance to systematically understand the electronic structure evolution during the reaction condition by developing the in-situ techniques. The understanding of deactivation mechanisms of single atom catalysts may guide the development of effective methods for stabilizing single atom catalysts.
(3) The electronic structure of single atoms bonded with various elementals. It has been reported that the doping of heteroatoms such as S or P can effectively tune the catalytic performance of M-N4 catalysts. Although various types of single atoms have been developed by several methods, it is still a great challenge to rational design the neighboring atoms of the single atom sites. For the single atoms on carbon-based supports, the replacement of C with other elementals can further tune electronic structure of single atom sites, thus improve their catalytic performance.
There are no conflicts to declare.
Xuebin Qiao: Writing – original draft, Investigation, Data curation. Lei Zhang: Writing – review & editing, Writing – original draft, Project administration, Investigation, Formal analysis, Conceptualization.
This work was financially supported by the National Natural Science Foundation of China (Nos. 22472101 and 22075203), Guangdong Science and Technology Department Program (No. 2021QN02L252) and Research Team Cultivation Program of Shenzhen University (No. 2023QNT007).
L. Zhang, K. Doyle-Davis, X. Sun, Energy Environ. Sci. 12 (2019) 492–517. doi: 10.1039/c8ee02939c
Z.X. Cai, Z.L. Wang, J. Kim, Y. Yamauchi, Adv. Mater. 31 (2019) e1804903. doi: 10.1002/adma.201804903
J. Kim, H.E. Kim, H. Lee, ChemSusChem 11 (2018) 104–113. doi: 10.1002/cssc.201701306
H. Xiang, W. Feng, Y. Chen, Adv. Mater. 32 (2020) e1905994. doi: 10.1002/adma.201905994
K. Jiang, S. Siahrostami, A.J. Akey, et al., Chem 3 (2017) 950–960. doi: 10.1016/j.chempr.2017.09.014
X. Li, W. Bi, M. Chen, et al., J. Am. Chem. Soc. 139 (2017) 14889–14892. doi: 10.1021/jacs.7b09074
X.X. Wang, D.A. Cullen, Y.T. Pan, et al., Adv. Mater. 30 (2018) 1706758. doi: 10.1002/adma.201706758
S. Fang, X. Zhu, X. Liu, et al., Nat. Commun. 11 (2020) 1029. doi: 10.1038/s41467-020-14848-2
C. Genovese, M.E. Schuster, E.K. Gibson, et al., Nat. Commun. 9 (2018) 935. doi: 10.1038/s41467-018-03138-7
M.J. Hulsey, B. Zhang, Z. Ma, et al., Nat. Commun. 10 (2019) 1330. doi: 10.1038/s41467-019-09188-9
X. Li, X. Yang, J. Zhang, Y. Huang, B. Liu, ACS Catal. 9 (2019) 2521–2531. doi: 10.1021/acscatal.8b04937
M. Yoo, Y.S. Yu, H. Ha, et al., Energy Environ. Sci. 13 (2020) 1231–1239. doi: 10.1039/c9ee03492g
Y. Pan, Y. Chen, K. Wu, et al., Nat. Commun. 10 (2019) 4290. doi: 10.1038/s41467-019-12362-8
N. Cheng, S. Stambula, D. Wang, et al., Nat. Commun. 7 (2016) 13638. doi: 10.1038/ncomms13638
P.N. Duchesne, Z.Y. Li, C.P. Deming, et al., Nat. Mater. 17 (2018) 1033–1039. doi: 10.1038/s41563-018-0167-5
M.T. Greiner, T.E. Jones, S. Beeg, et al., Nat. Chem. 10 (2018) 1008–1015. doi: 10.1038/s41557-018-0125-5
R. Lang, W. Xi, J.C. Liu, et al., Nat. Commun. 10 (2019) 234. doi: 10.1038/s41467-018-08136-3
J.C. Liu, Y.G. Wang, J. Li, J. Am. Chem. Soc. 139 (2017) 6190–6199. doi: 10.1021/jacs.7b01602
F. Dvorak, M.F. Camellone, A. Tovt, et al., Nat. Commun. 7 (2016) 10801. doi: 10.1038/ncomms10801
G. Spezzati, Y. Su, J.P. Hofmann, et al., ACS Catal. 7 (2017) 6887–6891. doi: 10.1021/acscatal.7b02001
Q. Feng, S. Zhao, Y. Wang, et al., J. Am. Chem. Soc. 139 (2017) 7294–7301. doi: 10.1021/jacs.7b01471
X. Cao, Y. Ji, Y. Luo, J. Phys. Chem. C 119 (2015) 1016–1023. doi: 10.1021/jp508625b
Y. Tang, C. Asokan, M. Xu, et al., Nat. Commun. 10 (2019) 4488. doi: 10.1038/s41467-019-12461-6
Y. Lu, J. Wang, L. Yu, et al., Nat. Catal. 2 (2019) 149–156.
A.J. Therrien, A.J.R. Hensley, M.D. Marcinkowski, et al., Nat. Catal. 1 (2018) 192–198. doi: 10.1038/s41929-018-0028-2
H. Wei, K. Huang, D. Wang, et al., Nat. Commun. 8 (2017) 1490. doi: 10.1038/s41467-017-01521-4
T. Li, J. Liu, Y. Song, F. Wang, ACS Catal. 8 (2018) 8450–8458. doi: 10.1021/acscatal.8b02288
X. Zhang, J. Guo, P. Guan, et al., Nat. Commun. 4 (2013) 1924. doi: 10.1038/ncomms2929
X. Wang, Z. Chen, X. Zhao, et al., Angew. Chem. Int. Ed. 57 (2018) 1944–1948. doi: 10.1002/anie.201712451
F. Yang, P. Song, X. Liu, et al., Angew. Chem. Int. Ed. 57 (2018) 12303–12307. doi: 10.1002/anie.201805871
K. Jiang, S. Siahrostami, T. Zheng, et al., Energy Environ. Sci. 11 (2018) 893–903. doi: 10.1039/c7ee03245e
M. Xiao, H. Zhang, Y. Chen, et al., Nano Energy 46 (2018) 396–403. doi: 10.1016/j.nanoen.2018.02.025
H.B. Yang, S.F. Hung, S. Liu, et al., Nat. Energy 3 (2018) 140–147. doi: 10.1038/s41560-017-0078-8
J.L. Shi, X.J. Zhao, L.Y. Zhang, et al., J. Mater. Chem. A 5 (2017) 19316–19322. doi: 10.1039/C7TA05483A
Y.G. Wang, D. Mei, V.A. Glezakou, J. Li, R. Rousseau, Nat. Commun. 6 (2015) 6511. doi: 10.1038/ncomms7511
Y. Chen, S. Ji, Y. Wang, et al., Angew. Chem. Int. Ed. 56 (2017) 6937–6941. doi: 10.1002/anie.201702473
C. Zhao, X. Dai, T. Yao, et al., J. Am. Chem. Soc. 139 (2017) 8078–8081. doi: 10.1021/jacs.7b02736
S. Sun, G. Zhang, N. Gauquelin, et al., Sci. Rep. 3 (2013) 1775. doi: 10.1038/srep01775
M. Kuhn, T.K. Sham, Phys. Rev. B 49 (1994) 1647–1661.
Q. Wang, X. Huang, Z.L. Zhao, et al., J. Am. Chem. Soc. 142 (2020) 7425–7433. doi: 10.1021/jacs.9b12642
L. Zhang, Q. Wang, R. Si, et al., Small 17 (2021) 2004453. doi: 10.1002/smll.202004453
S. Stankov, T. Ślęzak, M. Zając, et al., Mössbauer Spectrosc. 1 (2013) 1–42. doi: 10.1002/9781118714614.ch01
W. Liu, L. Zhang, X. Liu, et al., J. Am. Chem. Soc. 139 (2017) 10790–10798. doi: 10.1021/jacs.7b05130
J. Wang, Z. Huang, W. Liu, et al., J. Am. Chem. Soc. 139 (2017) 17281–17284. doi: 10.1021/jacs.7b10385
L. Lin, W. Zhou, R. Gao, et al., Nature 544 (2017) 80–83. doi: 10.1038/nature21672
J. Liu, M. Jiao, L. Lu, et al., Nat. Commun. 8 (2017) 15938. doi: 10.1038/ncomms15938
Y. Qu, B. Chen, Z. Li, et al., J. Am. Chem. Soc. 141 (2019) 4505–4509. doi: 10.1021/jacs.8b09834
S. Yang, J. Kim, Y.J. Tak, A. Soon, H. Lee, Angew. Chem. Int. Ed. 55 (2016) 2058–2062. doi: 10.1002/anie.201509241
C.H. Choi, M. Kim, H.C. Kwon, et al., Nat. Commun. 7 (2016) 10922. doi: 10.1038/ncomms10922
H. Yan, H. Cheng, H. Yi, et al., J. Am. Chem. Soc. 137 (2015) 10484–10487. doi: 10.1021/jacs.5b06485
Y. Xiong, J. Dong, Z.Q. Huang, et al., Nat. Nanotechnol. 15 (2020) 390–397. doi: 10.1038/s41565-020-0665-x
X. Li, W. Bi, L. Zhang, et al., Adv. Mater. 28 (2016) 2427–2431. doi: 10.1002/adma.201505281
L. Zhang, R. Si, H. Liu, et al., Nat. Commun. 10 (2019) 4936. doi: 10.1038/s41467-019-12887-y
Z. Geng, Y. Liu, X. Kong, et al., Adv. Mater. 30 (2018) 1870301. doi: 10.1002/adma.201870301
H. Tao, C. Choi, L.X. Ding, et al., Chem 5 (2019) 204–214. doi: 10.1016/j.chempr.2018.10.007
J.N. Tiwari, A.M. Harzandi, M. Ha, et al., Adv. Energy Mater. 9 (2019) 1900931. doi: 10.1002/aenm.201900931
D. Wang, Q. Li, C. Han, Z. Xing, X. Yang, Appl. Catal. B 249 (2019) 91–97. doi: 10.1089/gtmb.2018.0195
M. Xiao, L. Gao, Y. Wang, et al., J. Am. Chem. Soc. 141 (2019) 19800–19806. doi: 10.1021/jacs.9b09234
S. Tian, Z. Wang, W. Gong, et al., J. Am. Chem. Soc. 140 (2018) 11161–11164. doi: 10.1021/jacs.8b06029
X. Wang, W. Chen, L. Zhang, et al., J. Am. Chem. Soc. 139 (2017) 9419–9422. doi: 10.1021/jacs.7b01686
S. Ji, Y. Chen, Q. Fu, et al., J. Am. Chem. Soc. 139 (2017) 9795–9798. doi: 10.1021/jacs.7b05018
S. Cao, H. Li, T. Tong, et al., Adv. Funct. Mater. 28 (2018) 1802169. doi: 10.1002/adfm.201802169
G.X. Pei, X.Y. Liu, X. Yang, et al., ACS Catal. 7 (2017) 1491–1500. doi: 10.1021/acscatal.6b03293
S. Wei, A. Li, J.C. Liu, et al., Nat. Nanotechnol. 13 (2018) 856–861. doi: 10.1038/s41565-018-0197-9
S. Zhou, L. Shang, Y. Zhao, et al., Adv. Mater. 31 (2019) e1900509. doi: 10.1002/adma.201900509
J. Zhang, Y. Zhao, C. Chen, et al., J. Am. Chem. Soc. 141 (2019) 20118–20126. doi: 10.1021/jacs.9b09352
W.C. Cheong, W. Yang, J. Zhang, et al., ACS Appl. Mater. Interfaces 11 (2019) 33819–33824. doi: 10.1021/acsami.9b09125
H. Zhang, H.T. Chung, D.A. Cullen, et al., Energy Environ. Sci. 12 (2019) 2548–2558. doi: 10.1039/c9ee00877b
R. Jiang, L. Li, T. Sheng, et al., J. Am. Chem. Soc. 140 (2018) 11594–11598. doi: 10.1021/jacs.8b07294
Q. Li, W. Chen, H. Xiao, et al., Adv. Mater. 30 (2018) e1800588. doi: 10.1002/adma.201800588
Y. Chen, S. Ji, S. Zhao, et al., Nat. Commun. 9 (2018) 5422. doi: 10.1038/s41467-018-07850-2
S. Ji, Y. Chen, Z. Zhang, et al., Nanoscale Horiz. 4 (2019) 902–906. doi: 10.1039/c9nh00036d
Y. He, S. Hwang, D.A. Cullen, et al., Energy Environ. Sci. 12 (2019) 250–260. doi: 10.1039/c8ee02694g
X. Wang, P. Li, Z. Li, et al., Chem. Commun. 55 (2019) 6563–6566. doi: 10.1039/c9cc01717h
J. Li, M. Chen, D.A. Cullen, et al., Nat. Catal. 1 (2018) 935–945. doi: 10.1038/s41929-018-0164-8
P. Song, M. Luo, X. Liu, et al., Adv. Funct. Mater. 27 (2017) 1700802. doi: 10.1002/adfm.201700802
B. Qiao, A. Wang, X. Yang, et al., Nat. Chem. 3 (2011) 634–641. doi: 10.1038/nchem.1095
H. Wei, X. Liu, A. Wang, et al., Nat. Commun. 5 (2014) 5634. doi: 10.1038/ncomms6634
Y. Zhao, H. Zhou, W. Chen, et al., J. Am. Chem. Soc. 141 (2019) 10590–10594. doi: 10.1021/jacs.9b03182
J. Lin, A. Wang, B. Qiao, et al., J. Am. Chem. Soc. 135 (2013) 15314–15317. doi: 10.1021/ja408574m
Y. Chen, S. Ji, W. Sun, et al., J. Am. Chem. Soc. 140 (2018) 7407–7410. doi: 10.1021/jacs.8b03121
M. Piernavieja-Hermida, Z. Lu, A. White, et al., Nanoscale 8 (2016) 15348–15356. doi: 10.1039/C6NR04403D
S. Tian, W. Gong, W. Chen, et al., ACS Catal. 9 (2019) 5223–5230. doi: 10.1021/acscatal.9b00322
P. Aich, H. Wei, B. Basan, et al., J. Phys. Chem. C 119 (2015) 18140–18148. doi: 10.1021/acs.jpcc.5b01357
J. He, T. Dou, S. Diao, et al., ACS Appl. Nano Mater. 6 (2023) 13543–13550. doi: 10.1021/acsanm.3c02193
P. Li, M. Wang, X. Duan, et al., Nat. Commun. 10 (2019) 1711. doi: 10.1007/s00192-018-3796-y
J. Zhang, J. Liu, L. Xi, et al., J. Am. Chem. Soc. 140 (2018) 3876–3879. doi: 10.1021/jacs.8b00752
H. Li, L. Wang, Y. Dai, et al., Nat. Nanotechnol. 13 (2018) 411–417. doi: 10.1038/s41565-018-0089-z
X. Zhang, M. Zhang, Y. Deng, et al., Nature 589 (2021) 396–401. doi: 10.1038/s41586-020-03130-6
J. Zhang, Y. Zhao, X. Guo, et al., Nat. Catal. 1 (2018) 985–992. doi: 10.1038/s41929-018-0195-1
J. Ge, D. He, W. Chen, et al., J. Am. Chem. Soc. 138 (2016) 13850–13853. doi: 10.1021/jacs.6b09246
Y. Yao, S. Hu, W. Chen, et al., Nat. Catal. 2 (2019) 304–313. doi: 10.1038/s41929-019-0246-2
G.X. Pei, X.Y. Liu, A. Wang, et al., ACS Catal. 5 (2015) 3717–3725. doi: 10.1021/acscatal.5b00700
X. Zhang, G. Cui, H. Feng, et al., Nat. Commun. 10 (2019) 5812. doi: 10.1038/s41467-019-13685-2
F.R. Lucci, J. Liu, M.D. Marcinkowski, et al., Nat. Commun. 6 (2015) 8550. doi: 10.1038/ncomms9550
L. Zhang, H. Liu, S. Liu, et al., ACS Catal. 9 (2019) 9350–9358. doi: 10.1021/acscatal.9b01677
H. Wang, Q. Luo, W. Liu, et al., Nat. Commun. 10 (2019) 4998. doi: 10.1038/s41467-019-12993-x
F. Li, X. Liu, Z. Chen, Small Methods 3 (2019) 1800480. doi: 10.1002/smtd.201800480
Y. Li, H. Su, S.H. Chan, Q. Sun, ACS Catal. 5 (2015) 6658–6664. doi: 10.1021/acscatal.5b01165
Z. Li, H. He, H. Cao, et al., Appl. Catal. B 240 (2019) 112–121. doi: 10.1016/j.apcatb.2018.08.074
H. Shen, Y. Li, Q. Sun, J. Phys. Chem. C 121 (2017) 3963–3969. doi: 10.1021/acs.jpcc.7b00317
J. Zhao, J. Zhao, F. Li, Z. Chen, J. Phys. Chem. C 122 (2018) 19712–19721. doi: 10.1021/acs.jpcc.8b06494
S. Tian, Q. Fu, W. Chen, et al., Nat. Commun. 9 (2018) 2353. doi: 10.1038/s41467-018-04845-x
H. Yan, Y. Lin, H. Wu, et al., Nat. Commun. 8 (2017) 1070. doi: 10.1038/s41467-017-01259-z
Z. Lu, B. Wang, Y. Hu, et al., Angew. Chem. Int. Ed. 58 (2019) 2622–2626. doi: 10.1002/anie.201810175
J. Wang, W. Liu, G. Luo, et al., Energy Environ. Sci. 11 (2018) 3375–3379. doi: 10.1039/c8ee02656d
W. Ren, X. Tan, W. Yang, et al., Angew. Chem. Int. Ed. 58 (2019) 6972–6976. doi: 10.1002/anie.201901575
W. Zhu, L. Zhang, S. Liu, et al., Angew. Chem. Int. Ed. 59 (2020) 12664–12668. doi: 10.1002/anie.201916218
L. Cao, W. Liu, Q. Luo, et al., Nature 565 (2019) 631–635. doi: 10.1038/s41586-018-0869-5
B. Johannessen, Z.S. Hussain, D.R. East, M.A. Gibson, J. Phys. Conf. Ser. 430 (2013) 012119. doi: 10.1088/1742-6596/430/1/012119
Z. Jiang, W. Sun, H. Shang, et al., Energy Environ. Sci. 12 (2019) 3508–3514. doi: 10.1039/c9ee02974e
K. Nakatsuka, T. Yoshii, Y. Kuwahara, K. Mori, H. Yamashita, Phys. Chem. Chem. Phys. 19 (2017) 4967–4974. doi: 10.1039/C6CP06388H
Z. Zhang, J. Xiao, X.J. Chen, et al., Angew. Chem. Int. Ed. 57 (2018) 16339–16342. doi: 10.1002/anie.201808593
A. Zitolo, N. Ranjbar-Sahraie, T. Mineva, et al., Nat. Commun. 8 (2017) 957. doi: 10.1038/s41467-017-01100-7

Figure 1 The theoretical insights, reaction mechanism, characterization methods and atom/support/reactants effects for determining the electronic structure of single atoms.

Figure 2 (a) Density of states (DOS) for the metal center, atoms in the first coordinate shell and oxygen in Fe=O species. (b) Calculated charge density differences of Fe=O on Fe-NxCy SAs/N—C catalysts. Reproduced with permission [13]. Copyright 2019, Nature Publishing Group.

Figure 3 Optimized structures and energy profiles for Pt NP dispersion as isolated Pt atoms. (a, b) Calculated energies and surface structures for evaporation of a single Pt atom (Pt1) from a Pt(221) step (red value) or evaporation of a PtO2 species from an oxygen pre-covered Pt(221) step (black values). (c, d) Calculated energies and surface structures for dissociative capture of PtO2 over Fe2O3(0001) surface and concomitant formation of a Pt1 atom and evolved O2. Reproduced with permission [17]. Copyright 2019, Nature Publishing Group.

Figure 4 (a, b) The schematic, normalized XANES and FT-EXAFS spectra at Pt L3 edge of Pt single atoms on graphene obtained by ALD method. Reproduced with permission [14]. Copyright 2013, Nature Publishing Group. (c) XANES and FT-EXAFS spectra confirm the atomic dispersion of Co atoms in Co-N2, Co-N3, and Co-N4. Reproduced with permission [29]. Copyright 2018, Wiley-VCH.

Figure 5 (a) Curve-resolved XPS of the Ir 4f region for IrO2 and Ir SAs-NiO. (b) Curve-resolved XPS of the Ni 2p region for NiO and IrSA-NiO. Reproduced with permission [40]. Copyright 2020, American Chemical Society. (c) 57Fe Mössbauer spectra of the Fe−N−C-600/700/800 samples. Reproduced with permission [43]. Copyright 2017, American Chemical Society. (d) Experimental 57Fe Mössbauer transmission spectra measured for (Fe, Co)/N—C, and fittings with spectral components. Reproduced with permission [44]. Copyright 2017, American Chemical Society.

Figure 6 (a) R-space spectra from FT-EXAFS and proposed atomistic structure of the Pt/HSC. Reproduced with permission [49]. Copyright 2016, Nature Publishing Group. (b) XANES, FT-EXAFS spectra at the Pd K-edge and the schematic models of Pd1/graphene. Reproduced with permission [50]. Copyright 2019, American Chemical Society. (c) Structural characterization of Rh single atoms on N-doped carbon by XAFS spectroscopy. Reproduced with permission [51]. Copyright 2020, Nature Publishing Group.

Figure 7 (a-c) The XANES and FT-EXAFS R space fitting curves of Fe single atoms. Reproduced with permission [36]. Copyright 2017, Wiley-VCH. (d) The different pyrolysis strategy of Fe and Co-SAs/NSC. (e) The normalized XANES spectra and the FT-EXAFS spectra at Co K-edge of Co-SAs/NSC. Reproduced with permission [66]. Copyright 2019, American Chemical Society.

Figure 8 (a, b) The normalized XANES and FT-EXAFS spectra of Pt single atoms on FeOx at the Pt L3 edge. Reproduced with permission [77]. Copyright 2013, Nature Publishing Group. (c) Illustration of preparation process of positively charged Pt single atoms on TiO2. (d, e) AC—HAADF-STEM image and EXAFS R space spectra of Pt single atoms on TiO2. Reproduced with permission [81]. Copyright 2018, American Chemical Society. (f) The Au L3-edge XANES spectra of the experimental and simulated results for Au/NiFe LDH. (g) Proposed reaction pathways for Au/NiFe LDH. (h) Au L3-edge XANES spectra for Au/NiFe LDH. (i) Differential charge densities of NiFe LDH with and without Au atom when one O atom is adsorbed on the Fe site. Reproduced with permission [87]. Copyright 2018, American Chemical Society.

Figure 9 (a, b) The XANES and EXAFS R space curves of Pt single atoms on octahedral Pd surface. Reproduced with permission [96]. Copyright 2019, American Chemical Society. (c) Schematic illustration of synthesis of Pd1—Ni/SiO2, DRIFTS CO chemisorption of the Pd-Ni/SiO2 samples at the CO saturation coverage. and in situ XPS spectra of Pd-Ni/SiO2 samples and a Pd/SiO2 reference in the Pd 3d region. Reproduced with permission [97]. Copyright 2019, Nature Publishing Group.

Figure 10 X-ray absorption studies of the Pt-Ru dimers, Pt single atoms and Pt foil. (a, b) The normalized XANES spectra at the Pt L3-edge and L2-edge of the Pt-Ru dimers, Pt single atoms and Pt foil. (c) corresponding K2-weighted magnitude of Fourier transform spectra from EXAFS of the Pt-Ru dimers, Pt single atoms and Pt foil. (d) The normalized XANES spectra at the Ru K-edge and (e) corresponding K2-weighted magnitude of Fourier transform spectra from EXAFS of the Pt-Ru dimers and Ru metal. (f) The atom structures of different H adsorption on Pt-Ru dimer along with the adsorption energies. Reproduced with permission [53]. Copyright 2019, Nature Publishing Group.

Figure 11 (a-c) In situ XAFS measurements of the 1cFe–Pt/SiO2 catalyst at the Fe K edge and the Pt L3 edge for as-prepared 1cFe–Pt/SiO2–O, reduction at room temperature (1cFe–Pt/SiO2–R) and the PROX reaction at room temperature (1cFe–Pt/SiO2–P). (d) Proposed reaction pathways for CO oxidation on Fe1(OH)3@Pt(100). Reproduced with permission [109]. Copyright 2019, Nature Publishing Group. (e) In-situ W 4f7/2 XPS spectra of the catalyst under different gas atmospheres and temperatures. (f) Key steps of the proposed reaction mechanism based on in situ spectroscopy. Reproduced with permission [10]. Copyright 2019, Nature Publishing Group.

Figure 12 (a) Schematic of the homemade electrochemical in situ cell setup. (b) Cu K-edge XANES spectra of Cu-SA/SNC at different potentials during the ORR. (c) Normalized difference spectra for Cu K-edge XANES under in situ conditions. (d) The average oxidation states of Cu from XANES spectra. (e) k3-weighted FT-EXAFS spectra at ex situ, 1.0, 0.893 and 0.7 V vs. RHE. (f) The proposed ORR mechanism for the Cu-SA/SNC. Reproduced with permission [111]. Copyright 2019, RSC Publishing Group. Operando XANES spectra taken in 0.5 mol/L H2SO4 for (g) Co single atoms and (h) Fe single atoms. Reproduced with permission [114]. Copyright 2017, Nature Publishing Group.

Figure 13 Operando X-ray absorption spectroscopy and photoelectron spectroscopy. (a, b) Normalized operando Ni K-edge XANES spectra and Fourier transform magnitudes of EXAFS spectra of A-Ni-NG under open-circuit voltage bias in Ar (OCV-Ar) and CO2 (OCV-CO2), and at − 0.7 V (versus RHE). (c) Change in the XPS O 1s intensity induced by CO2 adsorption for A-Ni-NG. (d) Valence band spectra of A-Ni-NG before and after CO2 gas exposure, and after desorption of CO2 by thermal treatment at 500 ℃ for 20 min in vacuum. (e) The schematic shows the activation processes for CO2 molecules on the Ni(ⅰ) site. Reproduced with permission [33]. Copyright 2018, Nature Publishing Group. (f) Projected density of states on crucial structures of different metals from *CO and *COOH adsorption. (g) The initial condition of CoPc, adsorbed CO2, formed *COOH and adsorbed *CO on CoPc in presence of a hydronium in the water layer. Reproduced with permission [113]. Copyright 2019, Wiley-VCH.

Figure 14 (a-c) XANES and FT-EXAFS spectra at different applied voltages from the open-circuit condition to −0.07 V during HER, and the XANES data of the reference standards of Pt foil and PtO2. (d) Calculated DOS of Pt d orbital of clean surfaces, H 1s of hydrogen adsorbed on Pt sites and 5d of Pt sites with adsorbed hydrogen (denoted *Pt d orbital), from left to right, respectively. (e) Schematic DOS illustration of the interaction between Pt and H, the H 1s states split into bonding and antibonding states. Reproduced with permission [8]. Copyright 2020, Nature Publishing Group.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
 下载:
下载: