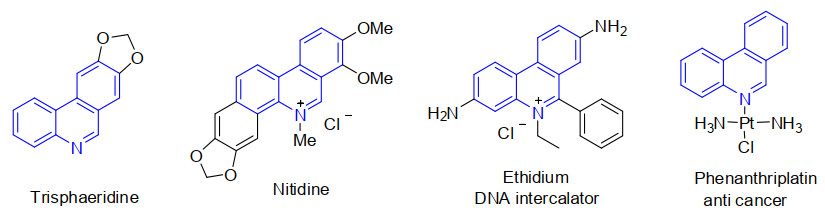
Figure 1.
Selected phenanthridine derivatives
Synthesis and Crystal Structure of 6-Cyclohexyl-8-methylphenanthridine
Zai-Gang LUO , Dan-Dan WEI , Cheng-Tong PENG , Chen-Fu LIU , Qi-Hua CHEN , Rong-Hao ZHANG , Xue-Mei XU

Phenanthridines, one kind of nitrogen-containing heterocycles existing widely in many natural products, have attracted great attention due to their significant applications in organic materials and pharmaceuticals[1-3]. The nitrogencontaining scaffolds of phenanthridine show various important biological activities such as antibacterial, antitumoral, DNA intercalator and anticancer properties (Fig. 1)[4-6]. Therefore, a great number of routes for the synthesis of phenanthridines have been described in the last several decades, such as the classic Pictet-Hubert reactions[7], transition metal-catalyzed reactions[8, 9], radical cascade reactions[10, 11], and other protocols[12-15]. Although these reported protocols have made a significant progress to synthesize these nitrogen-containing heterocycles, the development of an efficient, facile, and environmentally friendly method for the preparation of phenanthridines is still desired.
During the last three decades, various small-molecule HIV-1 integrase inhibitors have been reported[16, 17]. Especially, Ethidium-Arginine conjugates owning potent anti-HIV-1 activity at micromolar concentration without toxicity was reported by Cabrol-Bass's group in 1999[18]. And also, several kinds of HIV-1 integrase inhibitors have been previously reported by our group[19-21]. In continuation of our interest in obtaining new molecular entities with potential biological activities, we used phenanthridine skeleton as the platform to design a new kind of integrase inhibitors. Herein, we report the synthesis of 6-cyclohexyl-8-methylphenanthridine (IV) through four-step reactions by using 2-bromoaniline and 4-methylphenylboronic acid as the starting materials (Scheme 1). The fourth-step reaction, cyclization, was catalyzed by economical and environmentally benign iron and involves the use of di-tert-butyl peroxide (DTBP) as oxidant. The structure of the title compound was confirmed via 1H NMR, 13C NMR and HRMS. Meanwhile, the crystal structure of compound IV was determined by X-ray single-crystal diffraction analysis. The inhibition to the strand transfer process of HIV-1 integrase of the title compound was also evaluated.
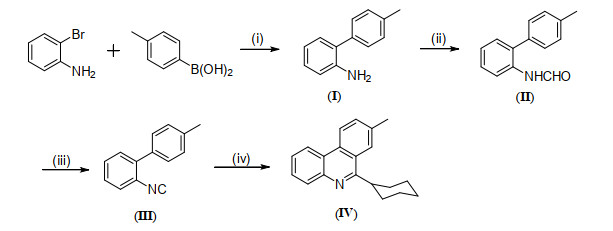
Reagents and conditions: (i) K2CO3, PdCl2(PPh3)2, ethylene glycol dimethyl ether, 80 ℃, 24 h; (ii) HCOOH, Ac2O, THF, rt, 2 h; (iii) POCl3, NEt3, THF, 0 ℃, 2 h; (iv) cyclohexanecarboxaldehyde, FeSO4, DTBP, PhCl, 130 ℃, 11 h
The melting point was measured on a SGW X-4 monocular microscope melting point apparatus with thermometer unadjusted. 1H NMR and 13C NMR spectra were acquired on a 400 MHz Bruker Avance spectrometer with CDCl3 as solvent. HRMS were obtained by ESI on a TOF mass analyzer. X-ray diffraction was performed using a Bruker Smart Apex CCD diffractometer. Unless otherwise noted, all materials were obtained from commercial suppliers and purified by standard procedures. Column chromatography was performed with silica gel (200~300 mesh, Qingdao Haiyang Chemical Co., Ltd, China). Petroleum ether used for column-chromatography has a boiling range of 60~90 ℃.
Intermediates (I), (II) and 2-isocyanobiphenyl (III) were prepared according to the related literature[22].
General procedure for the synthesis of 6-cyclohexyl-8-methylphenanthridine (IV) is described as follows: A Schlenk tube equipped with a magnetic stirring bar was charged with 2-isocyanobiphenyl III (0.2 mmol), cyclohexanecarboxaldehyde (0.4 mmol), FeSO4 (0.02 mmol), DTBP (0.6 mmol) and PhCl (2 mL). Then the tube was sealed and the resulting mixture was heated to 130 ℃ for 11 h. After cooling, the solvent was diluted with water (5 mL) and extracted with dichloromethane (3 × 10 mL). The combined organic layers were dried over anhydrous Na2SO4 and concentrated by a rotary evaporator, and the residue was purified by column chromatography on silica gel (V(petroleum ether)/V(ethyl acetate) = 80:1, 0.6 > Rf > 0.4) to provide the desired product 6-cyclohexyl-8-methylphenanthridine (IV). White solid, 33 mg, m.p. 129~131℃, isolated yield 60%; 1H NMR (400 MHz, CDCl3) δ 8.53 (d, J = 8.4 Hz, 1H), 8.48 (d, J = 8.0 Hz, 1H), 8.10 (d, J = 8.0 Hz, 1H), 8.06 (s, 1H), 7.61~7.67 (m, 2H), 7.57 (t, J = 7.2 Hz, 1H), 3.56~3.62 (m, 1H), 2.61 (s, 3H), 1.82~2.07 (m, 7H), 1.41~1.63 (m, 3H). 13C NMR (100 MHz, CDCl3) δ 165.06, 143.59, 136.92, 131.67, 130.87, 129.87, 127.96, 126.07, 125.10, 124.90, 123.46, 122.53, 121.67, 114.35, 41.86, 32.32, 26.93, 26.37, 22.04; HRMS m/z: calcd. for C20H22N [M+H]+, 276.1752; found, 276.1750.
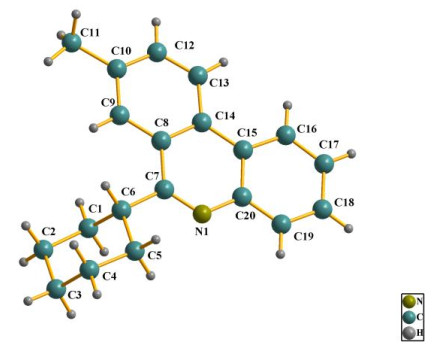
The X-ray crystallographic data of the colorless block crystal IV were collected and mounted on a glass fiber for measurement at room temperature. X-ray crystallographic data were collected at 240(2) K for IV. All measurements of the title compound were made on a Bruker Smart Apex CCD diffractometer equipped with graphite-monochromated CuKa radiation (λ = 1.54184 Å). The structure of compound IV was solved by direct methods, and then the non-hydrogen atoms were refined anisotropically with SHELXS-97 by applying a full-matrix least-squares procedure based on F2 values after they were located from the trial structure[23]. Moreover, the hydrogen atom positions were fixed geometrically at calculated distances. At the same time, they were allowed to ride on the parent atoms. For compound IV, a total of 7447 reflections were selected in the range of 3.62≤θ≤66.01º (h: −48~47, k: −30~36, l: −4~5) by using a ψ-ω scan mode, of which 2200 were independent with R = 0.2058 and 1233 were observed with I > 2σ(I), respectively. The final refinement showed R = 0.1285, wR = 0.2589 (w = 1/[σ2(Fo2) + (0.0000P)2 + 183.1230P], where P = (Fo2 + 2Fc2)/3) with GOF = 1.050, (Δρ)max = 0.433, (Δρ)min = –0.412 e/Å3 and (∆/σ)max = 0.001. Data were collected by Rapid Auto program. The hydrogen atoms bound to carbon were calculated theoretically. And the non-hydrogen atoms were located from the trial structure and then refined anisotropically with SHELXL-97 by using a full-matrix least-squares procedure based on F2 values[24-26]. The representative bond lengths and bond angles for compound IV are illustrated in Table 1.
 DownLoad:
CSV
DownLoad:
CSV
| Bond | Dist. | Angle | (º) | |
| N(1)–C(7) | 1.288(8) | C(7)–N(1)–C(20) | 122.3(6) | |
| N(1)–C(20) | 1.369(8) | N(1)–C(7)–C(6) | 120.1(6) | |
| C(1)–C(2) | 1.550(9) | C(19)–C(20)–N(1) | 120.0(6) | |
| C(1)–C(6) | 1.502(9) | C(1)–C(6)–C(5) | 109.3(6) | |
| C(2)–C(3) | 1.527(10) | C(4)–C(5)–C(6) | 108.0(6) | |
| C(5)–C(6) | 1.564(9) | C(3)–C(4)–C(5) | 111.2(7) | |
| C(6)–C(7) | 1.491(9) | C(2)–C(3)–C(4) | 109.3(6) | |
| C(7)–C(8) C(14)–C(15) |
1.515(9) 1.422(9) |
C(14)–C(15)–C(16) N(1)–C(7)–C(8) |
124.0(6) 121.3(6) |
The inhibition effect of compound IV was measured by HIV-1 integrase strand transfer activity assay, which was carried out as described previously with some minor modifications[20, 27]. Compounds diluted in DMSO were pre-incubated with 800 ng of integrase at 37.8 ℃ in the reaction buffer in the absence of Mn2+ for 10 min. Subsequently, 1.5 pmol of donor DNA and 9 pmol of target DNA were added and the reaction was initiated by the addition of 10 mmol/L Mn2+ into the final reaction volume. The reactions were carried out at 37.8 ℃ for 1 h and subsequent detection procedure was applied to detect the assay signals. Integrase inhibitor, baicalein, was used as the control compound (positive control), whereas no compound but only DMSO in the reaction mixture was set as the drug-free control (negative control). Based on the measured positive and negative values, the inhibition ratio of compound IV was calculated according to the calculation formula[27] as listed below (Scheme 2).
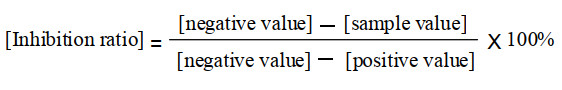
The final cyclization was conducted by testing different iron catalysts, oxidants and solvents (Table 2). FeSO4 was more efficient than other iron salts such as Fe(NO3)2 and FeCl2 (Table 2, entries 1~3). Replacing Fe(II) with Fe(III), such as FeCl3 Fe(NO3)3, Fe2(SO4)3 and Fe(acac)3, led to inferior results (Table 2, entries 4~7). Screening followed by other oxidants revealed that dicumyl peroxide (DCP), tert-butyl hydroperoxide (TBHP) and benzoyl peroxide (BPO) all met little success in this cyclization (Table 2, entries 8~10). Next, the solvents, including PhF, PhCF3 and PhCl provided the highest yield (Table 2, entries 2, 11, 12). Finally, the yield of the target product significantly increased to 60% when 3.0 equiv. of DTBP was added to the reaction system (Table 2, entry 13).
DownLoad:
CSV
| Entrya | Catalyst | Oxidant | Solvent | Yield (%)b |
| 1 | Fe(NO3)2 | DTBP | PhCl | trace |
| 2 | FeSO4 | DTBP | PhCl | 43 |
| 3 | FeCl2 | DTBP | PhCl | 21 |
| 4 | FeCl3 | DTBP | PhCl | 16 |
| 5 | Fe(NO3)3 | DTBP | PhCl | trace |
| 6 | Fe2(SO4)3 | DTBP | PhCl | 15 |
| 7 | Fe(acac)3 | DTBP | PhCl | 30 |
| 8 | FeSO4 | DCP | PhCl | 24 |
| 9 | FeSO4 | TBHP | PhCl | 25 |
| 10 | FeSO4 | BPO | PhCl | N.R. |
| 11 | FeSO4 | DTBP | PhF | 32 |
| 12 | FeSO4 | DTBP | PhCH3 | 37 |
| 13c | FeSO4 | DTBP | PhCl | 60 |
| aUnless otherwise specified, all reactions were carried out with 0.2 mmol of III, 0.4 mmol of cyclohexanecarbaldehyde, 10 mol% Fe catalyst and 2.0 equiv. oxidant in 2 mL solvent at 130 ℃ and reacted for 11 h in air. bIsolated yield. c3.0 equiv. oxidant. N.R. = No reaction | ||||
The structure for the target compound was confirmed by 1H NMR, 13C NMR and HRMS. For example, the 1H NMR spectrum of 6-cyclohexyl-8-methylphenanthridine IV shows that the aromatic hydrogen signals appeared at 7.57~8.53 ppm as four doublets, one multiplet and one singlet. The signals of cyclohexyl group were also well displayed at 3.56~1.82 ppm as three multiplets. And the signal as singlet at 2.61 ppm is due to the methyl group. The 13C NMR signals of the title compound possess the normal value. The HRMS spectrum of IV shows molecular ion peaks and [M+H]+ at m/z 276.1750. Furthermore, the solid of the target compound was recrystallized from dichloromethane/ethanol to give colorless and stable crystal suitable for single-crystal X-ray diffraction.
X-ray diffraction of the molecular structure of 6-cy-clohexyl-8-methylphenanthridine IV is demonstrated in Fig. 2. The tricyclic skeleton of phenanthridine is almost coplanar, with the maximum shift of the conjugated system of C(14) atom relative to C(10) to be 1.820°. Furthermore, the bond lengths and bond angles of the molecule are within normal limits. Obviously, the cyclohexyl group, connected with C7 of the molecular frame structure, maintains the thermodynamically stable chair conformation. Besides, the phenanthridine skeleton and the cyclohexyl group are approximately perpendicular to each other. However, the intermolecular and intramolecular hydrogen bonds are not found in the molecules.
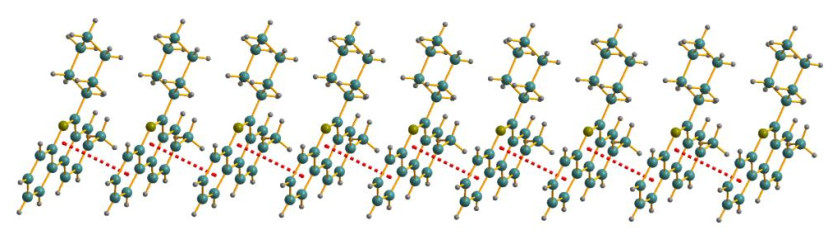
In addition, the crystal packing of IV illustrated in Fig. 3 discloses that the overall packing has stratified parallel arrangement. Moreover, one kind of π-π interaction between the two phenyl rings of the adjacent molecules at upper and lower levels can be viewed, which possessed a one-dimen-sional interaction model. As can be detected by the packing diagram with stratified arrangement, the centroid and vertical distances between the upper and lower arranges are 3.710 and 3.4479 Å, respectively. The dihedral angel of the adjacent tricyclic skeleton of phenanthridine is 1.122°.
Compound IV and positive control compound baicalein were tested against purified integrase and the data are concluded in Table 3.
DownLoad:
CSV
As listed in Table 3, the value of inhibition ratio of compound IV is 20.65% at the concentration of 50 μM. It means that the title compound has poor inhibitory activity towards HIV integrase. The reason maybe correlates with the rigid tricyclic skeleton or the bulky steric hindrance of the cyclohexyl group blocking the interaction of the molecule with HIV integrase. The further work based on the structure is in progress.
In summary, a convenient and efficient protocol for the synthesis of 6-cyclohexyl-8-methylphenanthridine was reported. The structure of the title compound was confirmed by 1H NMR, 13C NMR and HRMS. The X-ray diffraction of the molecular structure showed that the skeleton of phenanthridine is coplanar, and the cyclohexyl group maintains the chair conformation. A one-dimensional interaction model of the title compound was formed by one kind of π-π interactions between the two phenyl rings of the adjacent molecules at upper and lower levels. The anti-HIV integrase assay disclosed that the target compound has poor inhibitory activity. The mechanism study and applications of these nitrogen-containing heterocycles are underway in our lab.
Nakanishi, T.; Suzuki, M.; Saimoto, A.; Kabasawa, T. Structural considerations of NK109, an antitumor benzo[c]phenanthridine alkaloid. J. Nat. Prod. 1999, 62, 864–867. doi: 10.1021/np990005d
Abdel-Halim, O. B.; Morikawa, T.; Ando, S.; Matsuda, H.; Yoshikawa, M. New crinine-type alkaloids with inhibitory effect on induction of inducible nitric oxide synthase from crinum yemense. J. Nat. Prod. 2004, 67, 1119–1124. doi: 10.1021/np030529k
Sripada, L.; Teske, J. A.; Deiters, A. Phenanthridine synthesis via [2+2+2] cyclotrimerization reactions. Org. Biomol. Chem. 2008, 6, 263–265. doi: 10.1039/B716519F
Kanzawa, F.; Nishio, K.; Ishida, T.; Fukuda, M.; Kurokawa, H.; Fukumoto, H.; Nomoto, Y.; Fukuoka, K.; Bojanowski, K.; Saijo, N. Anti-tumour activities of a new benzo[c]phenanthridine agent, 2, 3-(methylenedioxy)-5-methyl-7-hydroxy-8-methoxybenzo[c]phenanthridinium hydrogensulphate dihydrate (NK109), against several drug-resistant human tumour cell lines. Br. J. Cancer. 1997, 76, 571–581. doi: 10.1038/bjc.1997.428
Ishikawa, T. Benzo[c]phenanthridine bases and their antituberculosis activity. Med. Res. Rev. 2001, 21, 61–72. doi: 10.1002/1098-1128(200101)21:1<61::AID-MED2>3.0.CO;2-F
Li, K.; Frankowski, K. J.; Belon, C. A.; Neuenswander, B.; Ndjomou, J.; Hanson, A. M.; Shanahan, M. A.; Schoenen, F. J.; Blagg, B. S. J.; Aubé, J.; Frick, D. N. Optimization of potent hepatitis C virus NS3 helicase inhibitors isolated from the yellow dyes thioflavine S and primuline. J. Med. Chem. 2012, 55, 3319–3330. doi: 10.1021/jm300021v
Pictet, A.; Hubert, A. Ueber eine neue synthese der phenanthridinbasen. Ber. Dtsch. Chem. Ges. 1896, 29, 1182–1189. doi: 10.1002/cber.18960290206
Grigg, R. D.; Van Hoveln, R.; Schomaker, J. M. Copper-catalyzed recycling of halogen activating groups via 1, 3-halogen migration. J. Am. Chem. Soc. 2012, 134, 16131–16134. doi: 10.1021/ja306446m
Fan, J.; Li, L.; Zhang, J.; Xie, M. Expeditious synthesis of phenanthridines through a Pd/MnO2-mediated C–H arylation/oxidative annulation cascade from aldehydes, aryl iodides and amino acids. Chem. Commun. 2020, 56, 2775–2778. doi: 10.1039/D0CC00300J
Jiang, H.; An, X.; Tong, K.; Zheng, T.; Zhang, Y.; Yu, S. Visible-light-promoted iminyl-radical formation from acyl oximes: a unified approach to pyridines, quinolines, and phenanthridines. Angew. Chem. Int. Ed. 2015, 54, 4055–4059. doi: 10.1002/anie.201411342
Zhou, Y.; Wu, C.; Dong, X.; Qu, J. Synthesis of 6-trichloromethylphenanthridines by transition metal-free radical cyclization of 2-isocyanobiphenyls. J. Org. Chem. 2016, 81, 5202–5208. doi: 10.1021/acs.joc.6b00885
Wang, L.; Zhu, H.; Guo, S.; Cheng, J.; Yu, J. T. TBHP-promoted sequential radical silylation and aromatisation of aryl isonitriles with silanes. Chem. Commun. 2014, 50, 10864–10867. doi: 10.1039/C4CC04773G
Fang, B.; Hou, J.; Tian, J.; Yu, W.; Chang, J. Synthesis of phenanthridines by I2-mediated sp3 C–H amination. Org. Biomol. Chem. 2020, 18, 3312–3323. doi: 10.1039/D0OB00433B
Li, G.; Kong, X.; Liang, Q.; Lin, L.; Yu, K.; Xu, B.; Chen, Q. Metal-free electrochemical coupling of vinyl azides: synthesis of phenanthridines and β-ketosulfones. Eur. J. Org. Chem. 2020, 6135–6145.
Zhang, B.; Studer, A. Recent advances in the synthesis of nitrogen heterocycles via radical cascade reactions using isonitriles as radical acceptors. Chem. Soc. Rev. 2015, 44, 3505–3521. doi: 10.1039/C5CS00083A
Dayam, R.; Deng, J.; Neamati, N. HIV-1 integrase inhibitors: 2003—2004 update. Med. Res. Rev. 2006, 26, 271−309. doi: 10.1002/med.20054
Dayam, R.; Gundla, R.; Al-Mawsawi, L. Q.; Neamti, N. HIV-1 integrase inhibitors: 2005—2006 update. Med. Res. Rev. 2008, 28, 118−154. doi: 10.1002/med.20116
Peytou, V.; Condom, R.; Patino, N.; Guedj, R.; Aubertin, A. M.; Gelus, N.; Bailly, C.; Terreux, R.; Cabrol-Bass, D. Synthesis and antiviral activity of Ethidium-Arginine conjugates directed against the TAR RNA of HIV-1. J. Med. Chem. 1999, 42, 4042–4053. doi: 10.1021/jm980728e
Luo, Z. G.; Zeng, C. C.; Yang, L. F.; He, H. Q.; Wang, C. X.; Hu, L. M. Synthesis of 6-sulfamoyl-4-oxoquinoline-3-carboxylic acid derivatives as integrase antagonists with anti-HIV activity. Chin. Chem. Lett. 2009, 20, 789–792. doi: 10.1016/j.cclet.2009.03.014
Luo, Z.; Zhao, Y.; Ma, C.; Li, Z.; Xu, X.; Hu, L.; Huang, N.; He, H. Synthesis, biological evaluation and molecular docking of calix[4]arene-based β-diketo derivatives as HIV-1 integrase inhibitors. Arch. Pharm. 2015, 348, 206–213. doi: 10.1002/ardp.201400390
Xu, X. M.; Luo, Z. G.; Han, X. X.; Liu, Q. N.; Li, R. Synthesis, crystal structure and biological activity of dimethyl 1-methyl-1, 4-dihydroquinoline-3, 4-dicarboxylate and tetramethyl pyrrolo[1, 2-a]quinoline-2, 3, 4, 5-tetracarboxylate. Chin. J. Struct. Chem. 2020, 39, 1271−1276.
Sha, W.; Yu, J. T.; Jiang, Y.; Yang, H.; Chen, J. The benzoyl peroxide-promoted functionalization of simple alkanes with 2-aryl phenyl isonitrile. Chem. Commun. 2014, 50, 9179−9181. doi: 10.1039/C4CC03304C
Bruker 2000, SMART (Version 5.0), SAINT-plus (Version 6), SHELXTL (Version 6.1), and SADABS (Version 2.03); Bruker AXS Inc. : Madison, WI.
Gu, S. J.; Qin, D. B.; Jin, L. H. Synthesis and crystal structure of calyx[4]arene bearing a 1, 8-bis(propoxy) anthracene-9, 10-dione. Chin. J. Struct. Chem. 2008, 27, 1035−1038.
Sheldrick, G. M. SHELXS-97, Program for X-ray Crystal Structure Solution. University of Göttingen, Germany 1997.
Sheldrick, G. M. SHELXL-97, Program for the Refinement of Crystal Structure. University of Göttingen: Göttingen, Germany 1997.
He, H. Q.; Ma, X. H.; Liu, B.; Chen, W. Z.; Wang, C. X.; Chen, S. H. A novel high-throughput format assay for HIV-1 integrase strand transfer reaction using magnetic beads. Acta Pharmacologica. Sinica 2008, 29, 397−404. doi: 10.1111/j.1745-7254.2008.00748.x

Scheme 1 Synthesis of 6-cyclohexyl-8-methylphenanthridine (IV)
Reagents and conditions: (i) K2CO3, PdCl2(PPh3)2, ethylene glycol dimethyl ether, 80 ℃, 24 h; (ii) HCOOH, Ac2O, THF, rt, 2 h; (iii) POCl3, NEt3, THF, 0 ℃, 2 h; (iv) cyclohexanecarboxaldehyde, FeSO4, DTBP, PhCl, 130 ℃, 11 h
Table 1. Selected Bond Lengths (Å) and Bond Angles (°) of Compound IV
| Bond | Dist. | Angle | (º) | |
| N(1)–C(7) | 1.288(8) | C(7)–N(1)–C(20) | 122.3(6) | |
| N(1)–C(20) | 1.369(8) | N(1)–C(7)–C(6) | 120.1(6) | |
| C(1)–C(2) | 1.550(9) | C(19)–C(20)–N(1) | 120.0(6) | |
| C(1)–C(6) | 1.502(9) | C(1)–C(6)–C(5) | 109.3(6) | |
| C(2)–C(3) | 1.527(10) | C(4)–C(5)–C(6) | 108.0(6) | |
| C(5)–C(6) | 1.564(9) | C(3)–C(4)–C(5) | 111.2(7) | |
| C(6)–C(7) | 1.491(9) | C(2)–C(3)–C(4) | 109.3(6) | |
| C(7)–C(8) C(14)–C(15) |
1.515(9) 1.422(9) |
C(14)–C(15)–C(16) N(1)–C(7)–C(8) |
124.0(6) 121.3(6) |
 下载: 导出CSV
下载: 导出CSV
Table 2. Optimization of Conditions for the Final Cyclization
| Entrya | Catalyst | Oxidant | Solvent | Yield (%)b |
| 1 | Fe(NO3)2 | DTBP | PhCl | trace |
| 2 | FeSO4 | DTBP | PhCl | 43 |
| 3 | FeCl2 | DTBP | PhCl | 21 |
| 4 | FeCl3 | DTBP | PhCl | 16 |
| 5 | Fe(NO3)3 | DTBP | PhCl | trace |
| 6 | Fe2(SO4)3 | DTBP | PhCl | 15 |
| 7 | Fe(acac)3 | DTBP | PhCl | 30 |
| 8 | FeSO4 | DCP | PhCl | 24 |
| 9 | FeSO4 | TBHP | PhCl | 25 |
| 10 | FeSO4 | BPO | PhCl | N.R. |
| 11 | FeSO4 | DTBP | PhF | 32 |
| 12 | FeSO4 | DTBP | PhCH3 | 37 |
| 13c | FeSO4 | DTBP | PhCl | 60 |
| aUnless otherwise specified, all reactions were carried out with 0.2 mmol of III, 0.4 mmol of cyclohexanecarbaldehyde, 10 mol% Fe catalyst and 2.0 equiv. oxidant in 2 mL solvent at 130 ℃ and reacted for 11 h in air. bIsolated yield. c3.0 equiv. oxidant. N.R. = No reaction | ||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们




 下载:
下载: