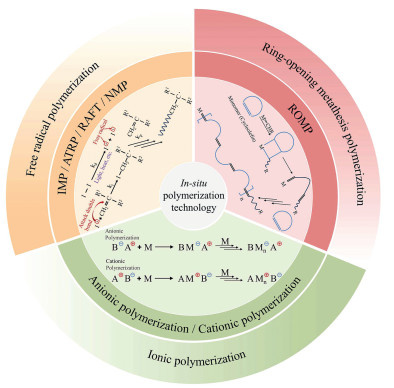
Figure 1.
Classification of in situ polymerization technology.
In-situ polymerization technology for all-solid state lithium batteries: Current status and future development
Zhaojun Chen , Yongqi Wang , Yongshun Liang , Liang Shan , Ying Wang , Jiyue Hou , Ziyi Zhu , Xue Li , Yiyong Zhang

Global energy demand growth and environmental protection requirements have driven research and development into high-performance, safe batteries [1]. Traditional graphite anodes have limited energy density improvement potential [2,3], so lithium metal anodes have become the focus of research into next-generation lithium metal batteries (LMBs) [4-6], due to their ultra-high theoretical specific capacity (3860 mAh/g) [7,8] and low redox potential of −3.04 V (relative to the standard hydrogen electrode) [9]. However, their commercialization still faces challenges [10,11]: Lithium dendrites may cause short circuits and thermal runaway [12-14], traditional organic electrolytes are flammable [15], and side reactions affect performance and lifespan [16,17].
To address the issues of lithium dendrites and electrolyte flammability, researchers have proposed using inorganic/polymer solid-state electrolytes to replace traditional electrolytes [18]. These materials offer advantages such as low flammability, high thermal stability, and leak-free safety [19], and can suppress lithium dendrite formation [20]. Among these, inorganic electrolytes, due to their non-flammability, high mechanical strength, and ionic conductivity, can enhance battery stability and safety [21], but their preparation is complex, they are costly, and they exhibit poor solid-solid interface contact [22,23]. In contrast, polymer electrolytes offer advantages such as low cost, good process compatibility [24], excellent interface contact [25], ease of processing [26], and design flexibility [27,28], making them a focus of recent attention [29].
Against this backdrop, in situ polymerization technology, as an innovative method for preparing solid-state electrolytes for all-solid-state lithium batteries, offers a new path for development [30]. This technology originates from the field of nanocomposites, where reactive monomers or prepolymers are directly filled into the nanolayer structure of the battery or the electrode surface, and then polymerized to form a solid-state electrolyte layer [31]. The specific implementation involves three steps: (1) Mixing reactive monomers (or soluble prepolymers) with initiators to prepare a precursor solution [32], (2) injecting the solution between the positive and negative electrodes to form a dispersed/continuous phase [33], and (3) the polymer solid-state electrolyte is in situ generated within the battery under temperature/pressure or light conditions [34]. Compared to traditional processes, this technology eliminates the complex coating-drying process, significantly reducing preparation costs [35]. Additionally, the polymerization process enhances the interface contact and compatibility between the electrolyte and electrodes, effectively improving the overall performance of the battery [36].
Based on its advantages, in situ polymerization has made significant progress in all-solid-state lithium batteries. Han et al. [37] utilized polymerization-induced phase separation (PIPS) technology to prepare plastic crystal elastic electrolytes (PCEE) using propyl acrylate (BA), butadiene nitrile (SN), and azobisisobutyronitrile (AIBN) to prepare a plastic crystal elastic electrolyte (PCEE), achieving simultaneous optimization of ionic conductivity (20 ℃, 10– 3 S/cm) and mechanical elasticity (elongation at break ≥300%) at the molecular level for the first time. Additionally, He et al. [38] replaced liquid electrolytes with in-situ polymerized acrylic ester-functionalized polyethylene glycol (PAP) quasi-solid-state electrolytes (QSPEs), forming a stable SEI layer rich in LiF on the lithium metal surface, thereby inhibiting the growth of dendritic crystals. Among these, the EDFA-based E-QSPE exhibits high oxidative stability (4.9 V) and is compatible with high-voltage NCM811 cathodes. This technology also facilitates structural design: Wang et al. [39] cross-linked modified metal-organic frameworks (Uio-66-MET) with poly(ethylene glycol) diacrylate (PEGDA) via a thiol-ene click reaction, forming a three-dimensional network electrolyte with -C-S-C- side chains, significantly enhancing tensile strength (9.4 MPa), elongation at break (500%), and ionic conductivity (24 times higher than pure PEGDA), while lowering the glass transition temperature (Tg, −56.7 ℃). Additionally, in situ polymerization enabled precise interface control. Zhang et al. [40] designed a trimethylolpropane-based electrolyte that can form a double-layer SEI (10–22 nm) in situ, with the inner layer enriched with LiF to enhance ion transport uniformity, and the outer layer containing lithium polyformaldehyde (LiPOM) to provide mechanical stability (dynamic modulus of 2.9 GPa), effectively suppressing SEI rupture. Zhou et al. [41] developed a P(VEC-IL) electrolyte based on 1-vinyl-3-ethylimidazole (VEIM), where the cations specifically adsorb on the NCM83 cathode surface to form a protective layer and promote the formation of anion-derived CEI, enhancing performance at high voltages (4.5 V) and wide temperature ranges (−30~70 ℃). Notably, Liu et al. [42] utilized this technology to construct a dual-interface synergistic regulation system (rich inorganic CEI layer and rich LiF gradient SEI layer), where additives and salts (LiBF4, LiTFSI) participate in forming the gradient SEI to suppress dendrites and side reactions, while initiators promote the formation of inorganic CEI at the cathode, significantly enhancing the performance of high-nickel solid-state lithium metal batteries. Currently, in situ polymerization has become one of the most commonly used methods for preparing solid-state electrolytes [43,44], and it is expected to make significant contributions to improving the performance of lithium metal batteries. Therefore, as shown in Fig. 1, this paper reviews three commonly used in situ polymerization methods in this technology, detailing their types, mechanisms, and applications to provide comprehensive guidance and reference, and discusses the challenges and prospects.
Although in situ polymerization technology has significantly advanced the development of all-solid-state lithium batteries, there remains a lack of systematic summaries of strategies based on polymerization mechanisms. In response, this paper reviews in situ polymerization methods for solid electrolytes, categorizing them into three types: radical polymerization, ring-opening migration polymerization, and ionic polymerization, based on mechanism differences. It also delves into the structure-property relationships between polymerization mechanisms and electrolyte performance, providing guidance for the design optimization of novel in situ polymerized electrolytes. Finally, the paper outlines the most promising research directions for this technology, aiming to guide the future development and commercialization of high-performance all-solid-state lithium batteries.
Free radical polymerization is a core technology in polymer synthesis, utilizing the reactivity of free radicals to initiate chain-growth addition polymerization reactions. Typically, olefin monomers containing unsaturated double bonds are used as raw materials, which undergo double bond cleavage followed by repeated addition reactions to form macromolecules. This method is the most commonly used technique for synthesizing organic polymer electrolytes.
Free radical polymerization is based on a chain reaction mechanism. First, the initiator decomposes upon heating or is excited by external energy (such as light, high-energy radiation, electrolysis, or plasma), producing highly reactive primary free radicals [45]. The primary free radicals rapidly add to olefin monomers (such as compounds containing double bonds), forming monomer free radicals (chain initiation) [46]. The active radical center continues to rapidly add to other monomer molecules, achieving chain growth. Chain growth can be terminated in the following ways: (1) Depletion of monomers, (2) addition of specific terminators, (3) the primary chain termination reaction, i.e., two growing chain radicals encountering each other and combining (forming saturated chains) or undergoing disproportionation reactions (generating saturated and unsaturated chains) [47].
The chain transfer phenomenon in polymerization reactions offers significant advantages for the design of polymer molecular chains. For example, strategies such as fixing anions to specific molecular chains [48] or introducing functional groups into the main chain [49], can effectively optimize the performance regulation of polymer solid-state electrolytes. Free radical polymerization systems exhibit significant controllability, with their in situ curing technology enabling interface integration between electrode materials and organic polymer electrolytes [50], and allowing adjustment of molecular weight and structure through parameters such as initiator concentration and reaction temperature [51]. Such polymerizations can be classified into multiple types based on their radical chain activity mechanisms, including Iniferter-mediated polymerization (IMP), atom transfer radical polymerization (ATRP), reversible addition-fragmentation chain transfer polymerization (RAFT), and nitrogen-oxygen radical-mediated polymerization (NMP). These methods collectively expand the strategic dimensions of polymer synthesis.
IMP is a unique and highly efficient radical polymerization method with broad application prospects. At its core is the initiator-transfer agent-terminator (Iniferter), which integrates initiation, transfer, and termination functions, serving as the key to achieving active polymerization. The process proceeds as follows: Iniferter decomposes under thermal, photochemical, or chemical conditions to generate free radicals that initiate monomer polymerization. During chain growth, it acts as a transfer agent to regulate polymer molecular weight and distribution, thereby enhancing reaction controllability. The IMP mechanism fundamentally involves a reversible equilibrium between growing chain free radicals and inert dormant species. Based on the initiation mechanism, they are primarily divided into two categories: (1) Thermally initiated (Thermoiniferter), such as tetraphenyldimethylsulfonyl (TPSN), which thermally decomposes to generate carbon-centered free radicals to initiate polymerization and terminate chain growth, forming dormant species containing initiator fragments [48], which can reversibly decompose to achieve active polymerization [49]; (2) photolytic (photoinitiators), primarily compounds containing diethyl dithiocarbonate imide groups. Under light exposure, these compounds decompose to produce two radicals: One initiates polymerization, while the other controls the process through transfer and termination [52]. Compared to traditional block copolymerization, this method involves fewer steps and achieves better results [53,54].
Previously, researchers developed high-performance polymer electrolytes using different polymerization strategies. In 2016, Porcarelli et al. [55] used 3 wt% AIBN as an initiator to thermally initiate the copolymerization of LiMTFSI, PEGM, and the crosslinking agent PEGDM in a PC plasticizer system, producing a crosslinked single-ion conductor polymer electrolyte (SIPE), which achieved high lithium-ion mobility (0.86), ionic conductivity (1.2 × 10⁻⁴ S/cm, which is 2–3 orders of magnitude higher than that of conventional systems), and a wide electrochemical window (5.0 V vs. Li+/Li). When applied to a LiFePO₄/Li battery, it provided a capacity of 126 mAh/g at 0.1 C and maintained a capacity retention rate of over 98% after 100 cycles, marking the first successful demonstration of efficient and stable operation of a room-temperature single-ion conductor (Fig. 2a). Subsequently, Cao et al. [56] in July 2017 utilized AIBN to thermally initiate the alternating copolymerization of SSPSILi and MA in DMF, synthesizing P(SSPSILi-alt-MA), which was then composite with PVDF-HFP to prepare a single-ion polymer electrolyte membrane; its room-temperature lithium-ion conductivity reached 2.67 mS/cm with a migration number of 0.98. The high dielectric constant of the acid anhydride groups promotes lithium-ion dissociation/distribution, while PVDF-HFP provides a continuous ionic pathway; A Li4Ti5O12/LiFePO4 full cell based on this electrolyte exhibits a capacity retention rate of 92.7% after 100 cycles at 0.5 C and a capacity of 36.9 mAh/g at 5 C, significantly outperforming liquid systems, effectively suppressing concentration polarization and lithium dendrites (Fig. 2b). Lin et al. [57] used AIBN to thermally initiate the in situ polymerization of liquid VEC monomers under vacuum at 80 ℃, preparing polyvinyl carbonate ethylene carbonate electrolyte (PVEC-PE), which exhibits an ionic conductivity of 2.1 mS/cm at 25 ℃, a wide electrochemical window up to 4.5 V (vs. Li+/Li), good interface compatibility, and ionic transport involving the coupling/decoupling of Li+ with C=O and C—O synergistic effects. When used in LiFePO₄/lithium metal batteries, the discharge capacity at 25 ℃/0.1 C is 165 mAh/g (104 mAh/g at −15 ℃), with capacities of 139 and 118 mAh/g after 100 cycles at 1 and 3 C rates, respectively. The lithium ion migration number increased to 0.40, and the interfacial resistance decreased to 70 Ω, enabling the development of high-performance polymer lithium metal batterie (Fig. 2c).
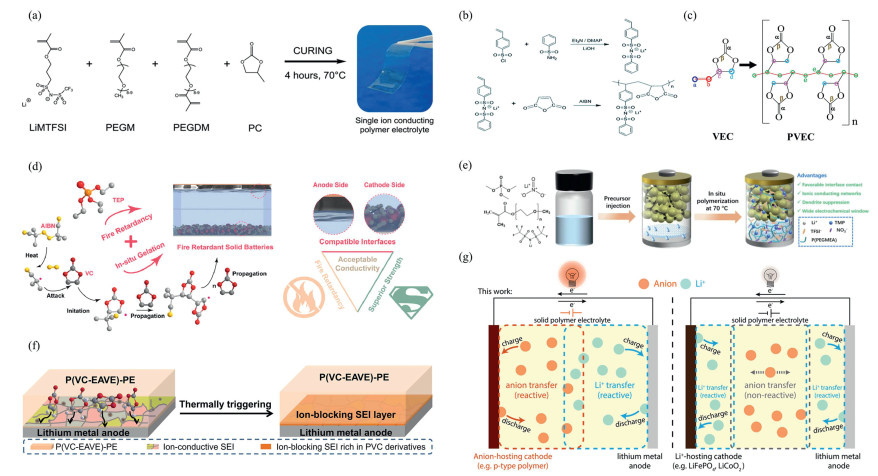
Moving into 2021, Tan et al. [58] developed a non-flammable solid electrolyte by encapsulating flame-retardant phosphates in a polycarbonate matrix using in situ curing technology. This electrolyte exhibits a high ionic conductivity of 4.4 mS/cm, a high Young's modulus of 12.4 GPa, a lithium ion mobility of 0.76, and a wide electrochemical window of 0–4.9 V, effectively suppressing lithium dendrite growth. The lithium symmetric battery assembled with this electrolyte stably cycled for 500 h at 0.5 mA/cm2 (overpotential 0.07 V). The Li||NCM811 full cell retained 87.7% of its capacity after 200 cycles, and the Li||Cu battery achieved a coulombic efficiency of 98.1%. Its safety was verified through a needle penetration test on an Ah-level pouch battery (Fig. 2d). From 2022 to 2024, in situ polymerization technology has shown diversified development: Zhang et al. [59] prepared an in situ radical polymerization electrolyte based on PEO, forming a complete ionic conductive network with excellent flexibility, and the pouch battery continued to function normally under bending and cutting conditions(Fig. 2e). Zhang et al. [60] designed a P(VC-EAVE)-based thermoresponsive polymer electrolyte that automatically shuts off ion channels when temperatures exceed limits (Fig. 2f). Sun et al. [61] used azobisisobutyronitrile as an initiator to prepare polyvinyl iron fluoride (PVF) with uniformly distributed iron, verifying its cycling stability and dendrite suppression capability (Fig. 2g). Meanwhile, Zhang et al. [62] developed an interface-compatible single-ion-conducting three-dimensional polymer electrolyte (3D-SIPE-LiFPA), significantly improving cycling life and safety (Fig. S1a in Supporting information). Han et al. [63] synthesized single-ion polymer electrolytes (SIPEs) using chain transfer initiators, achieving uniform lithium ion distribution and point-to-point migration, regulating ionic conductivity by three orders of magnitude, addressing wide-temperature-range cycling issues, and suppressing dendrite formation (Fig. S1b in Supporting information).
In 2024, multiple studies combined in situ polymerization technology with innovative concepts such as liquid metal composite materials and low-molecular-weight polymer confinement, significantly enhancing the performance and safety of lithium metal batteries. Specific advancements include: In March 2024, Peng et al. [64] proposed a strategy for designing solid polymer electrolytes through in situ initiation-transfer Iniferter-mediated polymerization, utilizing the weak coordination interactions of polymer chains to facilitate rapid lithium ion migration and constructing fast ion channels within oligomers (Fig. S1c in Supporting information); In April, Rui et al. [65] combined high-safety ethylene carbonate-free liquid electrolytes with in situ polymerization technology to prepare practical solid-state electrolytes that balance safety and electrochemical performance (Fig. S1d in Supporting information); Concurrently, Tong et al. [66] ddeveloped a quasi-solid composite electrolyte based on low-molecular-weight polyethylene glycol dimethyl ether (PEGDME), restricting it within the poly(methyl methacrylate) (PMMA) backbone via in situ Iniferter-mediated polymerization, offering a new approach for room-temperature solid-state lithium metal batteries (Fig. S1e in Supporting information); In July, Nguyen et al. [67] in South Korea combined an anode current collector coated with liquid metal@C nanocomposites with an in situ polymerized electrolyte, demonstrating the potential application of an anode-less battery (Fig. S1f in Supporting information).
Free radical-initiated in situ preparation technology is a key strategy for developing advanced battery materials, enabling efficient initiation and precise process control, promoting the formation of dense and uniform functional layers, thereby enhancing material interface bonding strength, structural stability, and overall electrochemical performance. By optimizing the initiation system and conducting in-depth studies on reaction kinetics, this technology holds promise for breakthroughs in the field of next-generation high-performance batteries. Although IMP technology offers mild reaction conditions, simple operation, system simplification, fewer steps, high polymerization degree, and excellent molecular weight control, enabling the preparation of polymers with narrower molecular weight distributions, it cannot achieve controlled synthesis and precise structural design of polymer electrolytes, which may lead to rapid degradation of electrochemical performance in battery applications.
ATRP is a novel controlled polymerization technique that combines the characteristics of radical polymerization and reactive polymerization. It offers the advantages of precise control over molecular weight (narrow distribution) and expanded monomer applicability (e.g., styrene, acrylates, vinyl derivatives), providing high flexibility for the preparation of functional polymers. Based on these characteristics, researchers have successfully synthesized various types of polymers using ATRP, including structurally defined homopolymers, random/alternating/step-growth/block-graft copolymers, dendritic polymers, and organic-inorganic hybrid materials, demonstrating broad cross-disciplinary application potential.
In the field of energy storage batteries, ATRP has even exerted its unique advantages. For example, Su et al. [68] synthesized fluoropolymers via ATRP and incorporated them into polyethylene glycol-based solid-state electrolytes, forming 21-arm fluoropolymers (21-β-CD-g-PTFEMA), which enhanced electrochemical window stability and conductivity, providing new insights for research on all-lithium and sodium metal batteries (Fig. 3a). Yang et al. [69] used ATRP to copolymerize 1,1,3-dioxolane with 1,3,5-triglycidyl isocyanurate (TGIC) in situ, resulting in a TPDOL electrolyte with excellent thermal stability and antioxidant properties, offering potential for intrinsically safe lithium metal batteries (Fig. 3b). Additionally, in terms of electrolyte structure optimization, Xie et al. [70] incorporated ether bonds with imidazolium ionic liquids into the main chain of multi-arm polymers via ATRP, preparing the tandem-structured S-P(PEGMA-IM) and parallel-structured P-P(PEGMA-IM). Electrochemical testing revealed that the SPEs derived from P-P(PEGMA-IM) exhibited higher ionic conductivity, lithium ion mobility, and stability compared to those from S-P(PEGMA-IM) (Fig. 3c). In membrane design, Zheng et al. [71] utilized ATRP to precisely construct nanoscale one-dimensional oriented Li+ flow, grafting the electron-negative poly(pentafluorostyrene propionate) (PPFPA) polymer onto a Celgard membrane, achieving rapid ion transport and ultra-stable lithium deposition (Fig. 3d).
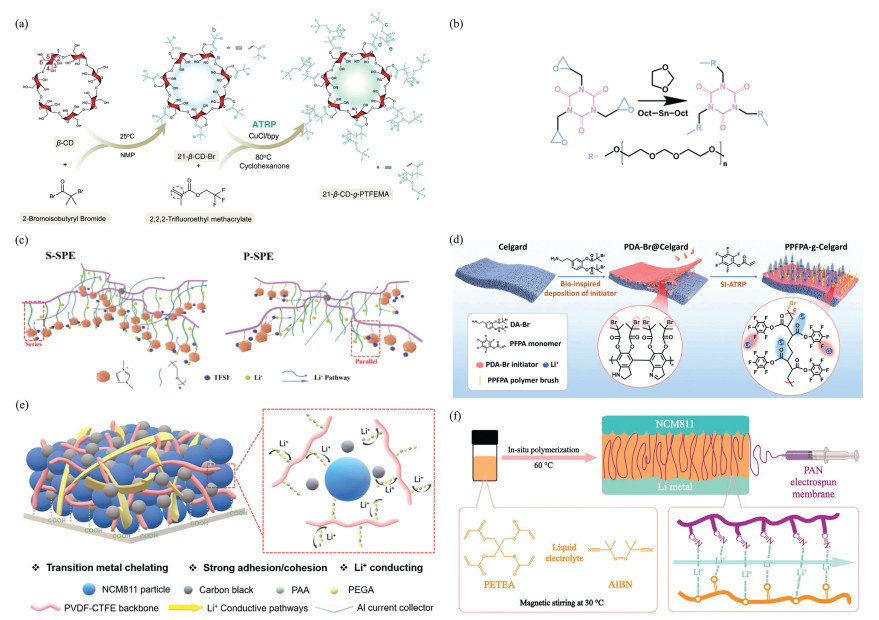
Meanwhile, ATRP has also made progress in adhesive modification: Liu et al. [72] used ATRP to covalently graft multifunctional poly(vinylidene fluoride-co-chlorotrifluoroethylene) (PVDF-CTFE) via ATRP to PVDF adhesives, yielding PVDF-CTFE-g-PEGA-co-PAA graft copolymers, which significantly enhanced flexibility, adhesive strength, and ionic conductivity (Fig. 3e). Shen et al. [73] used ATRP to prepare an in situ gel polymer electrolyte (PETEA-TCGG-PANZ) containing an electrolyte using pentylene glycol tetraacrylate (PETEA) as the monomer, investigated its polymerization mechanism and ionic transport properties, and provided a new design approach for in situ solid-state electrolytes for high-energy lithium metal batteries (Fig. 3f).
However, ATRP has the drawback of being sensitive to pH, which increases the difficulty of preparing polymer electrolytes and the complexity of reaction control, thereby hindering its application in energy storage batteries to some extent. Nevertheless, through optimizing reaction conditions and exploring new areas, ATRP still demonstrates significant potential and value. In summary, ATRP has been proven to be a precise, controllable, and flexible polymerization method, offering significant advantages and promising prospects in the fields of polymer electrolytes and functional polymer materials. Despite existing challenges, as research progresses, ATRP is expected to continue playing a unique role in driving technological advancements.
RAFT as an advanced active radical polymerization technology, provides an effective approach for synthesizing polymers with precise molecular weight and structure. Compared to ATRP, RAFT offers superior advantages in terms of monomer compatibility, oxygen tolerance, and molecular weight control, demonstrating greater commercial application potential. In its polymerization mechanism, disulfide ester derivatives act as chain transfer agents, reducing radical concentration through isomerization transfer reactions with growing chain radicals, thereby inhibiting irreversible diradical termination and enabling active polymerization. This process ensures that each polymer chain has nearly equal growth opportunities, ultimately forming a uniform and controllable polymer structure.
Based on the advantages of RAFT polymerization, several research teams have applied it to the synthesis of polymer solid-state electrolytes for lithium-metal batteries. Gao et al. [74] prepared a multi-grafted polymer network PPS (poly(dimethyl siloxane)-g-[poly(polyethylene glycol methyl ether methacrylate)-r-sodium poly(styrene sulfonate)]) via RAFT polymerization, demonstrating excellent electrochemical performance and mechanical strength (Fig. 4a). Guo et al. [75] prepared block copolymer electrolytes (BCPEs) via a one-step process combining RAFT polymerization and carboxylic acid-catalyzed ring-opening polymerization (ROP), enabling rapid Li+ migration in solid-state electrolytes (Fig. 4b). The following year, Guo et al. [76] utilized lithium-catalyzed ortho-polymerization reactions and RAFT reactions to in situ prepare hybrid, block, and brush-type solid-state polymer electrolytes, integrating the advantages of different matrices and demonstrating ideal mass transport performance and a broad electrochemical stability window (Fig. 4c). Hu et al. [77] modified nano-silica particles via RAFT polymerization and physical doping to prepare ionically bonded nano-particle-reinforced flexible composite polymer electrolytes, enhancing ionic conductivity and electrochemical stability. Zhou et al. [78] thermally initiated RAFT polymerization to synthesize dynamic borate-based self-healing polymer electrolytes in situ, exhibiting excellent mechanical properties and interface stability. Additionally, Zhan et al. [79] synthesized perfluorinated polyether derivatives (PPP) as electrolyte additives via RAFT polymerization, improving ionic transport efficiency and battery stability (Fig. 4d); Jin et al. [80] prepared amphiphilic ionic polymer interface protective layers, enhancing Li+ transport rates and battery cycling stability.
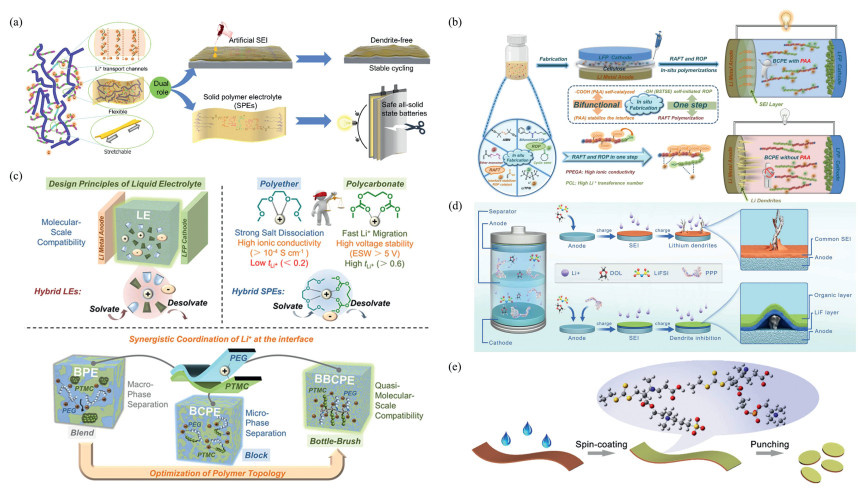
Makhlooghiazad et al. [81] combined sodium salts with ionic liquids via RAFT polymerization, demonstrating the feasibility of AB block copolymers as solid-state electrolytes for sodium batteries, and systematically investigated the properties of binary/ternary electrolyte systems. Kim et al. [82] employed a two-step process to develop a novel solid-state electrolyte: First, potassium persulfate was used as an initiator to prepare 20 nm cross-linked polystyrene (x-PS) core/poly(sodium styrene sulfonate) (PSS−Na+) shell core-shell structured nanoparticles via a 70 ℃ water-in-oil emulsion copolymerization reaction, followed by ion exchange to obtain the single-ion conductor PS (LiTFSI); Then, using ACVA as an initiator, a thiol-olefin click reaction at 75 ℃ was performed to graft vinyl polyethylene glycol (PEO) with a molecular weight of 2 kg/mol onto the surface of thiolized silica, forming 60 nm PEO-SiO₂ nanoparticles. The two particles were co-assembled in a 3:1 mass ratio to form an AB6 superlattice electrolyte. At 25 ℃, this material exhibited an ionic conductivity of 10−4 S/cm, a lithium ion mobility of 0.94, an electrochemical window of > 6 V, and an elastic modulus of 0.12 GPa, maintaining mechanical stability between 25 ℃ and 150 ℃. When applied to all-solid-state lithium-sulfur batteries, the first-cycle discharge capacity at 0.05 C reaches 1090 mAh/g, and the capacity at 3 C remains at 627 mAh/g. After 200 cycles, the capacity retention rate is 75%, significantly improving energy density, rate performance, and cycling stability.
Although RAFT polymerization technology has its advantages, its application still faces challenges: It requires the use of expensive high-purity specific catalysts/chain transfer agents, leading to complex synthesis and purification processes; and in actual operation, it is easily affected by interference, potentially deviating from the target narrow molecular weight distribution. To address these challenges, Sun et al. [83] developed a covalently bonded crosslinked network adhesive for Si/C electrodes using RAFT polymerization, demonstrating excellent cycling performance; Jiang et al. [84] synthesized a polymer adhesive (PAA) by copolymerizing acrylic monomers with a small amount of ether linkages through RAFT polymerization, effectively offsetting the capacity decay of Si-based electrodes (Fig. 4e).
RAFT polymerization, as an advanced reactive radical polymerization technology, has great potential in the synthesis of polymer solid-state electrolytes for lithium metal batteries. However, the polymerization conditions, catalyst and chain transfer agent selection, as well as the post-polymerization processing technology still need to be continuously optimized in order to fully leverage its advantages and overcome challenges.
NMP is an important controlled radical polymerization technique that utilizes stable nitroso oxide radicals to regulate the polymerization process [85]. This technology achieves precise control over polymer molecular weight and distribution by establishing a dynamic equilibrium between the active chain and the dormant state formed by the addition of nitrogen-oxygen radicals (Fig. 5a). Compared to ATRP and RAFT, the high chemical stability of NMP regulators confers tolerance to various functional groups, simplifying the synthesis of functional group-containing polymers and eliminating the need for purification and product odor, thereby driving the application of this technology. The advantages of NMP include high controllability, broad monomer applicability (e.g., styrene, acrylic esters), and mild reaction conditions (no strict anaerobic requirements), reducing operational complexity and costs. It enables efficient polymerization and produces polymers with narrow molecular weight distributions, which are critical for high-precision applications.
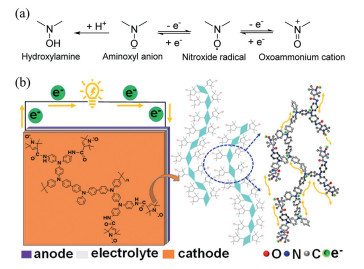
However, despite its advantages, NMP still faces significant challenges. First, its applicability is limited to a narrow range of monomers, restricting its scope of application. Second, limitations in molecular design make it difficult to meet the specific requirements of certain fields. Additionally, the synthesis and purification costs of nitrogen-oxygen radical regulators are high, hindering their large-scale industrial application. Furthermore, the polymerization rate of NMP may be slow, affecting the efficiency of large-scale production. More importantly, some NMPs are toxic and require special safety measures for handling, increasing application difficulty and costs. However, NMP still demonstrates significant potential in specific fields. For instance, Xiong et al. [86] synthesized a radical polymerization polymer P(DATPAPO-TPA) through in situ nitro oxide-mediated polymerization, featuring conjugated nitrogen-rich triphenylamine derivatives as the main chain and high-density nitrogen oxides as the side chains. This polymer exhibits high discharge capacity, high charge-discharge platforms, excellent rate performance, and good cyclability in aqueous zinc-ion batteries, validating the effectiveness of NMP in synthesizing high-performance polymers and providing new material options for the energy storage field (Fig. 5b).
Although nitrite-mediated polymerization has advantages, it also has challenges and limitations. In practical applications, its advantages and disadvantages, as well as factors such as cost and efficiency, must be weighed. With further research and technological advances, this method is expected to demonstrate unique advantages and application prospects in more fields in the future.
Free radical polymerization has garnered attention due to its simplicity, but its primary drawback lies in the difficulty of controlling polymer structure and molecular weight. In contrast, controlled free radical polymerization techniques (such as ATRP and RAFT) enable control over molecular weight and structure, but they require stringent and complex synthesis conditions. In recent years, transposition polymerization, particularly ROMP, has gained significant attention due to its ability to maintain the same composition as the monomer, automatically preserve the equimolarity of functional groups, and utilize mild polymerization conditions driven by ring tension.
ROMP is a chain-growth polymerization catalyzed by transition metal carbene complexes (such as Schrock molybdenum/tungsten or Grubbs ruthenium catalysts). Its core mechanism involves the isomerization of alkenes triggered by metal carbenes (e.g., Ru=CHR): the catalytic metal alkyl reacts with strained ring alkene monomers (e.g., cyclopentadiene or cyclooctene) via a [2 + 2] cycloaddition [87], forming a high-energy metal cyclobutane intermediate. This intermediate rapidly undergoes ring-opening, generating an active growing chain terminated with a metal alkyl group (polymer chain-Ru=CH-polymer chain), and small-molecule olefins are also produced during the cross-isomerization [88]. The newly formed polymeric metal alkyl carbocation then reacts with another monomer, repeating the [2 + 2] cycloaddition-ring-opening sequence. This cycle continues, with monomer molecules sequentially ring-opening and inserting between the metal alkyl carbocation and the growing chain. Polymerization termination is typically achieved through β-hydrogen elimination of the active chain, chain transfer agent reaction, or catalyst deactivation [89].
In 1973, Wright et al. [90] first used ROMP to prepare polyethylene oxide (PEO)-based solid-state electrolytes (SPE), demonstrating their potential. Advantages include: high monomer selectivity enhances SPE structural diversity; enables reversible deactivation radical polymerization (RDRP) to synthesize controllable high-performance biodegradable polymers; metal-free catalysis avoids current collector corrosion, polymerization without small molecule release, and molecular weight proportional to time, reducing interfacial side reactions.
In the field of lithium-ion batteries, ROMP technology has gained widespread application due to its advantages in the preparation of high-performance polymer electrolytes and the development of high-energy-density batteries. Specifically, Mu et al. [91] utilized surface-modified TiO2 nanoparticles as the heterogeneous catalytic center for ROMP, in situ synthesizing anti-agglomeration nanoparticle-hybrid crosslinked composite polymer electrolytes (NHCPEs), significantly enhancing ionic conductivity, lithium ion migration number, and thermal stability (Fig. S2a in Supporting information). Yang et al. [92] utilized ROMP to construct a LiF/LiCl/LiIn hybrid solid electrolyte interphase (SEI), achieving rapid Li+ transport and enhanced electronic insulation performance (Fig. S2b in Supporting information). For high-voltage solid-state batteries, Liu et al. [93] synthesized poly(DOX) polyethylene (PE) with excellent oxidation stability by regulating the molecular structure of the liquid precursor, providing long-cycle stability for high-voltage cathodes (Fig. S2c in Supporting information). Li et al. [94] ntroduced a multifunctional Lewis acid additive (THB) to simultaneously catalyze the in situ ROMP reaction of DOL and induce the formation of a fluorine/boron-enriched SEI layer. This strategy significantly suppressed lithium dendrite growth and extended the battery's cycle life (Fig. S2d in Supporting information). Li et al. [95] used ROMP to integrate polyethylene oxide electrolyte/composite electrodes, reducing interface resistance and improving stability, thereby optimizing the performance of lithium metal anodes (Fig. S2e in Supporting information). Hao et al. [96] prepared an ultra-adhesive interface electrolyte based on in situ polymerization of SnCl4. SnCl4 serves both as a polymerization initiator and a facilitator for the formation of the lithium metal alloy layer, exhibiting high ionic conductivity and electrochemical performance (Fig. S2f in Supporting information). Zheng et al. [97] introduced the bifunctional additive Sn(OTf)₂ into DOL-based electrolytes, in situ constructing a gel polymer electrolyte and Li-Sn alloy interface, resulting in a quasi-solid-state lithium metal battery with low internal resistance and strong interface compatibility. Sn2+ inhibits dendrite growth by improving electrolyte performance and promoting alloy layer formation (Fig. S2g in Supporting information). Tang et al. [98] developed a flame-retardant PDOL-TEP electrolyte for NCM811 lithium batteries, combining cycling stability and rate performance. The decomposition products of TEP can block combustion reactions, providing an effective solution for solid-state lithium metal batteries (Fig. S2h in Supporting information).
However, ROMP requires specific catalysts and strict conditions, increasing complexity and cost. Residual catalysts may affect electrolyte stability and battery performance. ROMP polymers may have poor compatibility with electrode interfaces, leading to high impedance. Therefore, when using ROMP to prepare electrolytes, it is necessary to weigh the pros and cons, optimize synthesis conditions, and select materials to achieve optimal performance. Although ring-opening isomerization polymerization holds great potential in lithium-ion batteries, it still faces challenges. Through continued research, it is hoped that more high-performance polymer electrolytes based on ROMP can be developed to drive the advancement of lithium-ion batteries.
In addition to known polymerization techniques, ion polymerization (including anionic and cationic polymerization) is a key addition polymerization technique for preparing polymer solid-state electrolytes (SSEs), primarily used in the lithium-ion battery field. By embedding migrating ions into polymer chains, this technique can construct efficient ion conduction pathways, enhancing battery safety and capacity. The resulting SSE exhibits excellent electrochemical stability, high ionic conductivity, and good mechanical toughness, enabling it to accommodate volume changes during charging and discharging. Its high flexibility allows precise control over polymer structure and properties, enabling customized design of lithium-ion batteries. This technology holds promise for driving the development of lithium-ion batteries toward higher performance and safety.
Ionic polymerization is a chain reaction in which charged ions (carbon anions/cations) serve as the active centers for chain growth. Monomers continuously add to ion pairs or free ions to achieve chain growth [99]. Based on the charge of the active center, it is divided into two categories: (a) Anionic polymerization: Strong nucleophilic initiators (e.g., RLi) attack monomers containing strong electron-withdrawing groups or conjugated systems (e.g., styrene, acrylonitrile), forming carbon anion centers. Strictly anhydrous, oxygen-free, and proton-free conditions (inert solvents) are required, exhibiting "active polymerization" characteristics (no spontaneous termination), with controllable molecular weight [100]. (b) Cationic polymerization: Electrophilic initiators/co-initiators (e.g., AlCl3/trace water) provide protons or carbocation centers, attacking monomers containing strong electron-donating groups (e.g., isobutylene, alkyl vinyl ethers), forming carbocation centers. A water-free environment (stable to oxygen) is required, typically conducted at low temperatures in polar solvents (e.g., chlorinated alkanes) to suppress chain transfer/termination, resulting in a broader molecular weight distribution [101]. Ionic polymerization is highly sensitive to the electron-withdrawing effects of monomer substituents [102]. The type of initiator determines the polymerization type; solvent polarity, temperature, and counterion properties influence the degree of ion dissociation, thereby regulating reaction rate, stereoregularity, and molecular weight distribution. Essentially, monomers add to the ionic active center via nucleophilic (anionic) or electrophilic (cationic) mechanisms [103].
Anionic polymerization is a key polymerization method that is highly dependent on initiators such as organic lithium metal compounds, alkali metals, amines, and Grignard reagents. The reaction must be carried out in a strictly anhydrous, oxygen-free inert atmosphere to avoid interference. The electron-withdrawing properties of the initiators cause them to act primarily on unsaturated monomers containing electron-withdrawing groups, and once the reaction is initiated, it continues until the monomers are depleted.
In the study of anionic polymerization, multiple teams have achieved significant results. Cui et al. [104] has developed a new strategy for in situ polymerization of PECA-GPE: Using lithium powder to trigger the rapid film formation of ethyl cyanoacrylate monomers in an electrolyte containing LiClO4. This electrolyte exhibits excellent performance (room temperature conductivity σ ~ 2.7 × 10−3 S/cm, t+ = 0.45, voltage window 4.8 V), with its cyano/ester groups strongly anchoring anions and forming continuous ionic channels. The innovative aspect of this method lies in utilizing the self-initiation properties of the monomer to achieve full electrode penetration, avoiding interface deactivation, thereby enabling LiFePO4 batteries based on PECA-GPE to exhibit bend-resistant performance and advancing the development of flexible lithium-ion batteries (Fig. 6a). Guo et al. [105] developed an electrolyte-mediated nano-confined strategy: LiTFSI/DMA was in situ solidified in LiCOF channels to form a DLC solid-state electrolyte. Its performance is outstanding: Li+ conductivity of 1.7 × 10⁻⁴ S/cm (a 100-fold increase), migration number of 0.85, ultra-thin (32 μm) and highly tough (tensile strength 22 MPa, elongation 49%). The DMA chains within the channels decouple strong interactions, "welding" to eliminate defects and form oriented channels, enabling the first foldable COF solid-state pouch battery (Fig. 6b). Zhou et al. [106] utilized LiI to catalyze the decarboxylation polymerization of VC, in situ preparing polyvinyl carbonate-LiI gel electrolyte. The resulting composite SEI containing Li₂CO₃ suppresses dendrite formation, combining high conductivity (1.8 × 10‒3 S/cm), excellent cycling performance (500 cycles at 5 mA/cm2), and self-shutoff safety features such as thermal-induced solidification at 80 ℃ (impedance increases by 103 times). This unique mechanism integrates stable lithium metal cycling with active thermal protection into a single system (Fig. 6c). Meisner et al. [107] utilized the discharge product Li2Sx from lithium-sulfur batteries as an initiator to in situ synthesize cross-linked solid-state polymer electrolytes via anionic ring-opening polymerization (AROP). This electrolyte is initiator-free, exhibits high ionic conductivity (6.9 mS/cm at 25 ℃), excellent cycling stability (overpotential < 0.25 V in a lithium symmetric battery after 400 h), and strong polysulfide shuttling inhibition capability (97.5% coulombic efficiency after 100 cycles), significantly enhancing the performance of lithium-sulfur batteries and providing an innovative strategy for solid-state electrolytes (Fig. 6d).
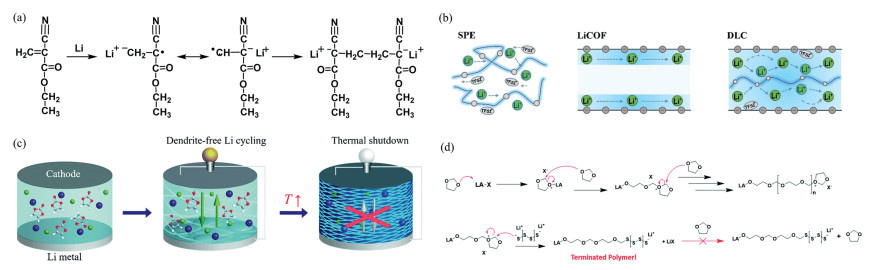
Anionic polymerization efficiently conducts ions through the polymer matrix by introducing a large number of mobile anions, significantly enhancing ionic conductivity. Carefully selecting monomers and polymerization conditions enables anionic polymers to form close interface contact with electrodes, reducing interface impedance. Additionally, adjusting polymer structure and composition can optimize mechanical properties to suit different battery requirements. However, anionic polymerization faces challenges: The reaction process is difficult to precisely control, prone to chain transfer and chain termination, leading to uneven polymer molecular weight and distribution; it is also sensitive to temperature and humidity, requiring strict control of the reaction environment. Furthermore, high-cost monomers and catalysts limit its large-scale application. Although initial performance is good, long-term stability still needs improvement. Anionic polymerization holds great potential in battery electrolytes but faces numerous challenges. Future research will focus on optimizing polymerization conditions, enhancing polymer stability, and reducing costs to drive broader application in battery technology.
Cationic polymerization is an important polymerization method in the field of battery electrolytes, initiated by electrophilic reagents (such as protonic acids, Lewis acids), and can be carried out under conditions ranging from room temperature to high temperature or high-energy radiation. The degree of polymerization depends on the activity of the initiator and monomer, and can be optimized by adjusting these parameters to enhance polymer performance.
This polymerization method has been applied to the preparation of high-performance gel polymer electrolytes (GPEs) and SPEs. Liu et al. [108] utilized LiPF6 to in situ initiate the cationic polymerization of 1,3-dioxane and 1,2-dimethoxyethane liquid electrolytes at room temperature, converting them into quasi-solid GPEs. The GPE monomer polymerization conversion rate reached 91.0%, with a polymer backbone melting point of 60 ℃, capable of withstanding temperature changes during battery formation, and reducing the contact resistance between the lithium-sulfur battery cathode and electrolyte, enabling rapid Li+ transport and blocking polysulfide migration (Fig. 7a). Similarly, Hwang et al. [109] prepared a room-temperature crosslinked GPE via in situ cationic polymerization of triethylene glycol diethylene ether (TEGDVE) with LiBF₄, exhibiting a wide electrochemical window, high ionic conductivity, and excellent electrochemical performance (Fig. 7b). Zhao et al. [110] prepared SPE that closely contacts all battery components by adding an aluminum-based initiator to trigger the in situ cationic ring-opening polymerization of molecular ether. This SPE exhibits high ionic conductivity, low interfacial resistance, and uniform lithium deposition at room temperature, demonstrating high coulombic efficiency and long-life battery potential (Fig. 7c).
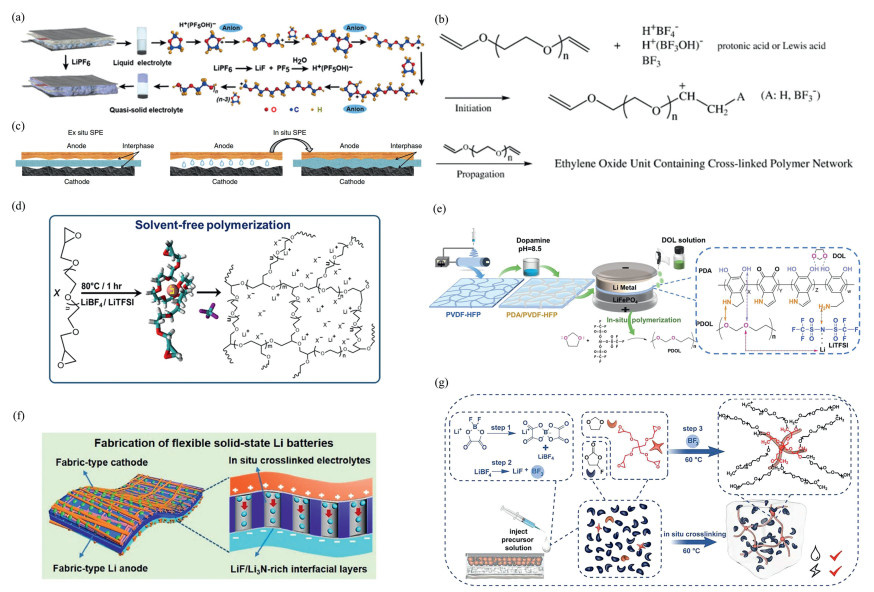
Nair et al. [111]. employed thermally induced cationic ring-opening polymerization (CROP) using polyethylene glycol diglycidyl ether (PEGDGE) as the monomer, combined with a LiBF4/LiTFSI double salt system (optimal composition P85A: 85 wt% PEGDGE, 5 wt% LiBF₄, 10 wt% LiTFSI), and cured under nitrogen at 80 ℃ for 1 h without a solvent, forming a cross-linked three-dimensional network solid polymer electrolyte (SPE). This SPE exhibits a low glass transition temperature (< −50 ℃), high room-temperature ionic conductivity (0.113 mS/cm, reaching 1 mS/cm at 70 ℃), and a wide electrochemical window (> 5.5 V vs. Li/Li+, reaching 7 V when LiBF4 dominates). Chen et al. [112] prepared self-reinforced gel polymer electrolytes (GPEs) by in situ ion polymerization of 1,3-dioxolane (DOL) on a nanofiber scaffold, achieving safe lithium metal batteries (LMBs) and good cycling stability (Fig. 7d). In addition, Xiang et al. [113] utilized multifunctional tri(pentafluorophenyl)borane (TB) additives to in situ form a flame-retardant polymer 1,3-dioxolane electrolyte (PDE), which provides a stable electrode-electrolyte interface and exhibits both excellent flame retardancy and oxidative stability (TB) additive. The electrolyte is formed in situ by multifunctional TB additive, which not only provides a stable electrode-electrolyte interface, but also possesses good flame retardancy and oxidative stability (Fig. 7e). Wen et al. [114] reported the design of ultra-thin crosslinked SPEs with high ionic conductivity, high mechanical strength, and rapid interfacial charge transfer at room temperature (Fig. 7f). Zhu et al. [115] prepared crosslinked gel polymer electrolytes (c-GPE) using tetraarm crosslinking agents via cationic ring-opening polymerization (CROP) crosslinking, which exhibit high solvent adsorption capacity, good oxidative stability, and excellent cycling performance (Fig. 7g).
Despite the challenges associated with ion polymerization technology, such as the difficulty in precisely controlling the polymerization process (prone to chain transfer/termination reactions, leading to structural and performance inconsistencies), sensitivity to temperature and humidity (requiring strict environmental control), and stringent catalyst selection (which may limit polymer diversity and performance), researchers have developed various strategies to address these issues (e.g., multifunctional initiators, optimized conditions, introduction of functional groups, and development of novel catalysts). These strategies have enhanced the controllability and efficiency of polymerization while expanding the performance and application potential of polymers. In the field of solid-state batteries, ion polymerization can be used to prepare polymer electrolytes with excellent ionic conductivity, mechanical properties, and thermal stability by regulating conditions and monomers, introducing functional groups, and optimizing catalysts, thereby meeting diverse requirements. In the future, the application prospects of this technology will be even more promising.
Lithium solid-state battery technology is mature and has broad application prospects [116]. In situ polymerization, as an innovative method, has garnered attention for simplifying processes, improving interface compatibility, and suppressing lithium dendrites [117,118]. This paper focuses on the progress of in situ curing technologies such as radical polymerization, ring-opening metathesis polymerization (ROMP), and cationic/anionic polymerization [119], emphasizing the critical importance of these methods for polymer molecular structure [120], ionic conductivity, and the commercialization of solid-state lithium batteries [121-123].
Different in-situ polymerization methods have distinct characteristics: Radical polymerization offers a wide range of monomer choices, fast reaction rates, and mature processes, but residual initiators/catalysts may degrade oxidative stability and cycle life, and some systems exhibit oxygen sensitivity and gas evolution issues. ROMP typically does not use metal catalysts, resulting in polyether electrolytes with high ionic conductivity, good interface compatibility, and thermal stability. However, monomer selection is limited (primarily to cyclic ethers, lactones, etc.), and precise control of polymerization rate/degree and curing shrinkage is required. Ion polymerization can produce polymers with a wide electrochemical window and high thermal stability (such as polyacrylates), but it is sensitive to water/impurities, has an uncontrollably fast reaction rate, and has limited monomers. The cost of the initiation system and residual effects also need to be considered. In terms of safety, ROMP (especially metal-free catalysis) is generally the best, with few byproducts, high thermal stability, and low gas generation. Free radical polymerization is highly influenced by residues and poses risks of thermal runaway and gas generation. Ion polymerization electrolytes have good thermal stability, but their high reactivity poses process challenges, and the potential toxicity of initiators must be addressed. A summary comparison table of core performance metrics for different polymerization methods is shown in Table S1 (Supporting information).
Although performance has improved in recent years through optimizing monomers, polymerization conditions, and introducing inorganic fillers, commercialization still faces specific limitations: The bottleneck of radical polymerization lies in oxidative decomposition caused by residues; ROMP is constrained by monomers, curing control, and temperature sensitivity; and ion polymerization is limited by harsh synthesis conditions, uncontrollable ultra-high reaction rates, high-cost initiator systems, and potential toxicity. Additionally, the technology still faces widespread challenges in terms of conductivity, stability, interfacial resistance, compatibility, cost, and scalable production [124,125].
Future prospects indicate that ROMP (especially those based on cyclic ether monomers) hold the most promising commercial potential in the mid-to-short term for power battery applications due to their superior comprehensive performance and safety potential. Radical polymerization (particularly controlled RAFT polymerization) may offer opportunities in specific applications or as a modification technique. Ion polymerization currently faces the greatest challenges in commercialization, with potential lying in systems requiring high-voltage stability. The cross-integration of these three methods is also an important trend. Ultimately, the dominant technology will depend on advancements in materials, processes, and cost control. In-situ polymerization technology, with its unique advantages in improving interface contact and suppressing dendrites, remains a key direction for future solid-state electrolyte research. Its core drawbacks (such as residual effects, limited monomer selection, process control challenges, and cost) require ongoing overcoming, while its core advantages (process simplification, interface optimization, and dendrite suppression potential) are the key drivers propelling further exploration. In-situ polymerization presents both opportunities and challenges in the design of next-generation high-performance lithium metal batteries:
(1) Development of liquid-free in situ polymerization technology: Design in situ polymerization technology based on flame-retardant plasticizers to eliminate liquid components (The core is to develop polymerizable monomers/oligomers containing flame-retardant groups such as phosphonitrile/phosphate esters, which directly form a solid network after polymerization); utilize AI to screen monomers that simultaneously exhibit high ionic conductivity, low flammability, and polymerization activity; explore the use of solid ionic conductors such as lithium salt crystals as "solid plasticizers" in the polymerization process.
(2) Optimize the correlation between polymerization process and performance: Conduct a systematic study on the effects of initiators (activity/dosage/half-life) and temperature on the electrochemical window of the electrolyte and room-temperature ionic conductivity; evaluate polymerization degree (molecular weight, polydispersity index PDI) using gel permeation chromatography (GPC) to balance performance; establish a machine learning predictive model linking polymerization parameters, polymer structure, and electrolyte performance; develop a high-throughput in situ characterization platform (combining rheology, EIS, and GPC) to monitor polymerization processes and performance evolution in real time.
(3) Construct efficient structures and study interfacial mechanisms: Combine 3D printing/directed freezing to prepare continuous porous frameworks (ceramic/polymer) and optimize in situ polymerization technology to construct structures that facilitate Li+ migration and suppress dendrite formation; design monomers with flexible segments or self-contracting compensation mechanisms to mitigate volume shrinkage; utilize in situ X-ray/neutron imaging, atomic force microscopy (AFM), and molecular dynamics (MD)/phase field simulations to elucidate electrode/electrolyte interface evolution and failure mechanisms, guiding interface modification (e.g., in situ formation of the SEI layer).
(4) Develop novel electrolytes and additives: Develop single-ion polymer electrolytes, MOF/COF/POM composite electrolytes, and multifunctional additives (Strategies include in situ polymerization for direct synthesis of single-ion conductor polymers (anion anchoring)); designing MOF/COF/POM particles with polymerizable functional groups to achieve strong interface bonding; developing multifunctional polymerizable additives that combine initiation, plasticization, flame retardancy, interface stabilization, or optimization of lithium solventization structure, utilizing AI to generate new molecular structures.
(5) Advancing industrialization and sustainability: Adopting infrared/laser/electromagnetic induction precise temperature control to address non-uniform heating; develop low-viscosity, high-wettability precursors or add surfactants to improve wettability; design self-venting/low-pressure processes to eliminate bubbles; optimize battery pack thermal management and simulate mass production using AI; strengthen green design (bio-based monomers) and recycling research, develop efficient recycling processes based on chemical depolymerization/physical separation, and achieve sustainability across the entire lifecycle.
To address challenges and adapt to market and competitive changes [126,127], future efforts should prioritize the development of new electrolyte materials, enhance interface stability, optimize cost control, and scale up production [128]. Continuous innovation is needed to drive the development of solid-state electrolyte and all-solid-state battery technologies and seize opportunities [129-131]. Therefore, strengthening industry-academia-research collaboration and promoting collaborative innovation are key to accelerating the development of the solid-state electrolyte industry [132,133].
The authors declare that they have no known competing financial interests or personal relationships that could have influenced the work reported in this paper.
Zhaojun Chen: Writing – original draft, Conceptualization. Yongqi Wang: Investigation. Yongshun Liang: Investigation. Liang Shan: Investigation. Ying Wang: Investigation. Jiyue Hou: Writing – review & editing. Ziyi Zhu: Funding acquisition. Xue Li: Supervision. Yiyong Zhang: Writing – review & editing.
This work was supported by the Yunnan Province Youth Talent Special Project (No. YNQR-QNRC-2020-011), the Scientific and Technological Project of Yunnan Precious Metals Laboratory (No. YPML-20240502015), the Natural Science Foundation of Yunnan Province (No. 202401AS070646), the Key Research and Development Program of Yunnan Province (No. 202403AA080018), the Yunnan Engineering Research Center Innovation Ability Construction and Enhancement Projects (No. 2023-XMDJ-00617107).
Supplementary material associated with this article can be found, in the online version, at doi:
Y. Bai, W. Ma, W. Dong, et al., ACS Appl. Mater. Interfaces 15 (2023) 26834–26842. doi: 10.1021/acsami.3c04234
J. Chai, Z. Liu, J. Ma, et al., Adv. Sci. 4 (2017) 1600377. doi: 10.1002/advs.201600377
J. Chai, Z. Liu, J. Zhang, et al., ACS Appl. Mater. Interfaces 9 (2017) 17897–17905. doi: 10.1021/acsami.7b02844
Y. Cui, X. Liang, J. Chai, et al., Adv. Sci. 4 (2017) 1700174. doi: 10.1002/advs.201700174
T. Dong, J. Zhang, G. Xu, et al., Energy Environ. Sci. 11 (2018) 1197–1203. doi: 10.1039/c7ee03365f
A. Du, H. Zhang, Z. Zhang, et al., Adv. Mater. 31 (2019) 1805930. doi: 10.1002/adma.201805930
F. Gambino, M. Gastaldi, A. Jouhara, et al., J. Power Source Adv. 30 (2024) 100160. doi: 10.1016/j.powera.2024.100160
D. Goonetilleke, N. Sharma, J. Kimpton, et al., Front. Energy Res. 6 (2018) 64. doi: 10.3389/fenrg.2018.00064
W. Han, J. Zheng, H. Huang, et al., J. Membr. Sci. 713 (2025) 123374. doi: 10.1016/j.memsci.2024.123374
X. He, J. Lin, G. Ge, et al., J. Power Sources 555 (2023) 232346. doi: 10.1016/j.jpowsour.2022.232346
J.K. Hu, H. Yuan, S.J. Yang, et al., J. Energy Chem. 71 (2022) 612–618. doi: 10.1016/j.jechem.2022.04.048
J. Hu, Y. Gao, S. Yang, et al., Adv. Funct. Mater. 34 (2024) 2311633. doi: 10.1002/adfm.202311633
I. Kim, S. Jang, K.H. Lee, Y. Tak, G. Lee, Energy Storage Mater. 40 (2021) 229–238. doi: 10.1016/j.ensm.2021.05.019
J. Kim, W. Lee, J. Seok, et al., J. Energy Chem. 66 (2022) 226–236. doi: 10.1016/j.jechem.2021.08.017
X. Li, C. Wang, W. Nan, et al., Mater. Today Chem. 40 (2024) 102219. doi: 10.1016/j.mtchem.2024.102219
Z. Li, H.X. Xie, X.Y. Zhang, X. Guo, J. Mater. Chem. A 8 (2020) 3892–3900. doi: 10.1039/c9ta09969g
C. Liu, F. Zhu, Z. Huang, et al., Chem. Eng. J. 434 (2022) 134644. doi: 10.1016/j.cej.2022.134644
Q. Lu, C. Wang, D. Bao, et al., Energy Environ. Sci. 6 (2023) e12447.
Y.Q. Mi, W. Deng, C. He, et al., Angew. Chem. Int. Ed. 62 (2023) e202218621. doi: 10.1002/anie.202218621
A. Murali, R. Ramesh, M. Sakar, S. Park, S.S. Han, RSC Adv. 14 (2024) 30618–30629. doi: 10.1039/d4ra05134c
L. Zhang, H. Gao, S. Xiao, et al., ACS Mater. Lett. 4 (2022) 1297–1305. doi: 10.1021/acsmaterialslett.2c00238
L. Nie, S. Chen, M. Zhang, et al., Nano Res. 17 (2024) 2687–2692. doi: 10.1007/s12274-023-6096-x
D. Shao, X. Wang, X. Li, et al., J. Solid State Electrochem. 23 (2019) 2785–2792. doi: 10.1007/s10008-019-04382-7
S.Z. Zhang, X.H. Xia, D. Xie, et al., J. Power Sources 409 (2019) 31–37. doi: 10.3847/1538-4365/aafb32
X. Yang, F. Su, M. Hou, et al., Dalton Trans. 50 (2021) 7041–7047. doi: 10.1039/d1dt00807b
C. Wang, K. Fu, S.P. Kammampata, et al., Chem. Rev. 120 (2020) 4257–4300. doi: 10.1021/acs.chemrev.9b00427
Z. Yao, Y. Kang, M. Hou, et al., Adv. Funct. Mater. 32 (2022) 2111919. doi: 10.1002/adfm.202111919
D. Zhang, K. Ye, Y. Yao, et al., Carbon 142 (2019) 278–284. doi: 10.1016/j.carbon.2018.10.062
Z. Wei, J. Huang, Z. Liao, et al., Energy Storage Mater. 72 (2024) 103714. doi: 10.1016/j.ensm.2024.103714
F. Wu, J. Maier, Y. Yu, Chem. Soc. Rev. 49 (2020) 1569–1614. doi: 10.1039/c7cs00863e
N. Wu, Y. Li, A. Dolocan, et al., Adv. Funct. Mater. 30 (2020) 2000831. doi: 10.1002/adfm.202000831
F. Wang, C.M. Yang, Y.Q. Wang, et al., Rare Met. 43 (2024) 1461–1487. doi: 10.1007/s12598-023-02486-8
F. Wang, R.W. Huang, W.C. Han, et al., Rare Met. 43 (2024) 3661–3676. doi: 10.1007/s12598-024-02704-x
C. Xu, K. Zhang, D. Zhang, et al., Nano Energy 68 (2020) 104318. doi: 10.1016/j.nanoen.2019.104318
P. Xu, X. Lin, Z. Sun, et al., J. Energy Chem. 72 (2022) 186–194. doi: 10.1016/j.jechem.2022.04.025
L. Yang, Y. Mu, L. Zou, et al., Nano Lett. 24 (2024) 13162–13171. doi: 10.1021/acs.nanolett.4c02894
J. Han, M.J. Lee, K. Lee, et al., Adv. Mater. 35 (2023) 2205194. doi: 10.1002/adma.202205194
Y. He, X. Shan, Y. Li, et al., Energy Storage Mater. 68 (2024) 103281. doi: 10.1016/j.ensm.2024.103281
H. Wang, Q. Wang, X. Cao, et al., Adv. Mater. 32 (2020) 2001259. doi: 10.1002/adma.202001259
Q.K. Zhang, X.Q. Zhang, J. Wan, et al., Nat. Energy 8 (2023) 725–735. doi: 10.1038/s41560-023-01275-y
Q. Zhou, H. Zhao, C. Fu, et al., Angew. Chem. Int. Ed. 63 (2024) e202402625. doi: 10.1002/anie.202402625
Q. Liu, Y. Sun, S. Wang, et al., Mater. Today 64 (2023) 21–30. doi: 10.1016/j.mattod.2023.02.011
D. Zhou, Y. He, R. Liu, et al., Adv. Energy Mater. 5 (2015) 1500353. doi: 10.1002/aenm.201500353
D. Zhang, Z. Xie, K. Zhang, et al., Chem. Eng. Sci. 241 (2021) 116695. doi: 10.1016/j.ces.2021.116695
K. Matyjaszewski, Adv. Mater. 30 (2018) 1706441. doi: 10.1002/adma.201706441
X. Pan, M. Fantin, F. Yuan, K. Matyjaszewski, Chem. Soc. Rev. 47 (2018) 5457–5490. doi: 10.1039/c8cs00259b
V. Vijayakumar, B. Anothumakkool, S. Kurungot, M. Winter, J.R. Nair, Energy Environ. Sci. 14 (2021) 2708–2788. doi: 10.1039/d0ee03527k
L. Zhao, Q. Dong, Y. Wang, et al., Angew. Chem. Int. Ed. 63 (2024) e202412280. doi: 10.1002/anie.202412280
M. Ma, F. Shao, P. Wen, et al., ACS Energy Lett. 6 (2021) 4255–4264. doi: 10.1021/acsenergylett.1c02036
P. Purohit, A. Bhatt, R.K. Mittal, M.H. Abdellattif, T.A. Farghaly, Front. Bioeng. Biotechnol. 10 (2023) 1044927. doi: 10.3389/fbioe.2022.1044927
C. Yu, J.K. Ha, M. Park, et al., Chem. Sci. 16 (2025) 11626–11636. doi: 10.1039/d5sc02594j
H. Gao, Y. Zhou, K. Wang, et al., Adv. Energy Mater. 15 (2025) 2501379. doi: 10.1002/aenm.202501379
R. Wang, W. Wang, Y. Zhang, et al., Angew. Chem. Int. Ed. 64 (2025) e202417605. doi: 10.1002/anie.202417605
M. Ma, X. Guo, P. Wen, et al., Angew. Chem. Int. Ed. 63 (2024) e202407304. doi: 10.1002/anie.202407304
L. Porcarelli, A.S. Shaplov, F. Bella, et al., ACS Energy Lett. 1 (2016) 678–682. doi: 10.1021/acsenergylett.6b00216
C. Cao, Y. Li, Y. Feng, et al., J. Mater. Chem. A 5 (2017) 22519–22526. doi: 10.1039/C7TA05787C
Z. Lin, X. Guo, Z. Wang, et al., Nano Energy 73 (2020) 104786. doi: 10.1016/j.nanoen.2020.104786
S.J. Tan, J. Yue, Y.F. Tian, et al., Energy Storage Mater. 39 (2021) 186–193. doi: 10.1016/j.ensm.2021.04.020
X. Zhang, M. Jia, Q. Zhang, et al., Chem. Eng. J. 448 (2022) 137743. doi: 10.1016/j.cej.2022.137743
H. Zhang, L. Huang, H. Xu, et al., eScience 2 (2022) 201–208. doi: 10.1016/j.esci.2022.03.001
Z. Sun, K. Xi, J. Chen, et al., Nat. Commun. 13 (2022) 3209. doi: 10.1007/s00170-021-08080-5
S. Zhang, F. Sun, X. Du, et al., Energy Environ. Sci. 16 (2023) 2591–2602. doi: 10.1039/d3ee00558e
S. Han, P. Wen, H. Wang, et al., Nat. Mater. 22 (2023) 1515–1522. doi: 10.1038/s41563-023-01693-z
H. Peng, T. Long, J. Peng, et al., Adv. Energy Mater. 14 (2024) 2400428. doi: 10.1002/aenm.202400428
X. Rui, R. Hua, D. Ren, et al., Adv. Mater. 36 (2024) 2402401. doi: 10.1002/adma.202402401
R. Tong, Y. Huang, C. Feng, Y. Dong, C. Wang, Adv. Funct. Mater. 34 (2024) 2315777. doi: 10.1002/adfm.202315777
M.H. Nguyen, D. Kim, B. Kim, S. Park, Adv. Funct. Mater. (2024) 2407179.
Y. Su, X. Rong, A. Gao, et al., Nat. Commun. 13 (2022) 4181. doi: 10.1038/s41467-022-31792-5
S. Yang, H. Yuan, N. Yao, et al., Adv. Mater. 36 (2024) 2405086. doi: 10.1002/adma.202405086
Z. Xie, Y. Zhou, C. Ling, et al., Chin. Chem. Lett. 33 (2022) 1407–1411. doi: 10.1016/j.cclet.2021.08.031
S. Zheng, L. Mo, K. Chen, et al., Adv. Funct. Mater. 32 (2022) 2201430. doi: 10.1002/adfm.202201430
T. Liu, R. Parekh, P. Mocny, et al., ACS Mater. Lett. 5 (2023) 2594–2603. doi: 10.1021/acsmaterialslett.3c00485
Z. Shen, J. Zhong, S. Jiang, et al., ACS Appl. Mater. Interfaces 14 (2022) 41022–41036. doi: 10.1021/acsami.2c11397
S. Gao, Z. Li, Z. Zhang, et al., Energy Storage Mater. 55 (2023) 214–224. doi: 10.1016/j.ensm.2022.11.049
K. Guo, J. Wang, Z. Shi, et al., Angew. Chem. Int. Ed. 62 (2023) e202213606. doi: 10.1002/anie.202213606
K. Guo, S. Li, J. Wang, et al., ACS Energy Lett. 9 (2024) 843–852. doi: 10.1021/acsenergylett.3c02422
J. Hu, W. Wang, B. Zhou, et al., J. Membr. Sci. 575 (2019) 200–208. doi: 10.1016/j.memsci.2019.01.025
B. Zhou, T. Deng, C. Yang, et al., Adv. Funct. Mater. 33 (2023) 2212005. doi: 10.1002/adfm.202212005
Y. Zhan, P. Zhai, T. Song, W. Yang, Y. Li, Chem. Eng. J. 491 (2024) 151974. doi: 10.1016/j.cej.2024.151974
T. Jin, M. Liu, K. Su, et al., ACS Appl. Mater. Interfaces 13 (2021) 57489–57496. doi: 10.1021/acsami.1c19479
F. Makhlooghiazad, L. Miguel Guerrero Mejía, G. Rollo-Walker, et al., J. Am. Chem. Soc. 146 (2024) 1992–2004. doi: 10.1021/jacs.3c10510
B. Kim, M.J. Park, Mater. Horiz. 10 (2023) 4139–4147. doi: 10.1039/d3mh00913k
C. Sun, H. Zhang, P. Mu, et al., ACS Nano 18 (2024) 2475–2484. doi: 10.1021/acsnano.3c11286
S. Jiang, B. Hu, Z. Shi, et al., Adv. Funct. Mater. 30 (2020) 1908558. doi: 10.1002/adfm.201908558
Y. Xie, K. Zhang, Y. Yamauchi, K. Oyaizu, Z. Jia, Mater. Horiz. 8 (2021) 803–829. doi: 10.1039/d0mh01391a
Y. Xiong, Z. Wang, Y. Li, Y. Chen, L. Dong, J. Am. Chem. Soc. 146 (2024) 22777–22786. doi: 10.1021/jacs.4c07941
M. Scholl, S. Ding, C.W. Lee, R.H. Grubbs, Org. Lett. 1 (1999) 953–956. doi: 10.1021/ol990909q
A.A. Nagarkar, A.F.M. Kilbinger, Nat. Chem. 10 (2018) 573–573. doi: 10.1038/s41557-018-0044-5
J. Xu, N. Hadjichristidis, Prog. Polym. Sci. 139 (2023) 101656. doi: 10.1016/j.progpolymsci.2023.101656
P.V. Wright, Electrochim. Acta 43 (1998) 1137–1143. doi: 10.1016/S0013-4686(97)10011-1
K. Mu, W. Dong, W. Xu, et al., Adv. Funct. Mater. (2024) 2405969.
T. Yang, W. Zhang, Y. Liu, et al., Small 19 (2023) 2303210. doi: 10.1002/smll.202303210
Y. Liu, H. Zou, Z. Huang, et al., Energy Environ. Sci. 16 (2023) 6110–6119. doi: 10.1039/d3ee02797j
T. Li, K. Chen, B. Yang, et al., Chem. Sci. 15 (2024) 12108–12117. doi: 10.1039/d4sc02010c
G. Li, K. Liang, Y. Li, et al., Nano Res. 17 (2024) 5216–5223. doi: 10.1007/s12274-024-6463-2
Q. Hao, Y. Gao, F. Chen, et al., Chem. Eng. J. 481 (2024) 148666. doi: 10.1016/j.cej.2024.148666
J. Zheng, W. Zhang, C. Huang, et al., Mater. Today Energy 26 (2022) 100984. doi: 10.1016/j.mtener.2022.100984
W. Tang, T. Zhou, Y. Duan, et al., Carbon Neutral. 3 (2024) 386–395. doi: 10.1002/cnl2.130
A.G. Nguyen, M.H. Lee, J. Kim, C.J. Park, Nano-Micro Lett. 16 (2024) 83. doi: 10.1007/s40820-023-01294-0
K. Mu, D. Wang, W. Dong, et al., Adv. Mater. 35 (2023) 2304686. doi: 10.1002/adma.202304686
S. Zou, Y. Yang, J. Wang, et al., Energy Environ. Sci. 17 (2024) 4426–4460. doi: 10.1039/d4ee00822g
S. Qin, Y. Yu, J. Zhang, et al., Adv. Energy Mater. 13 (2023) 2301470. doi: 10.1002/aenm.202301470
Y.Q. Mi, W. Deng, C. He, et al., Angew. Chem. 135 (2023) e202218621. doi: 10.1002/ange.202218621
Y. Cui, J. Chai, H. Du, et al., ACS Appl. Mater. Interfaces 9 (2017) 8737–8741. doi: 10.1021/acsami.6b16218
D. Guo, D.B. Shinde, W. Shin, et al., Adv. Mater. 34 (2022) 2201410. doi: 10.1002/adma.202201410
H. Zhou, H. Liu, Y. Li, et al., J. Mater. Chem. A 7 (2019) 16984–16991. doi: 10.1039/c9ta02341k
Q.J. Meisner, S. Jiang, P. Cao, et al., J. Mater. Chem. A 9 (2021) 25927–25933. doi: 10.1039/d1ta08244b
F.Q. Liu, W.P. Wang, Y.X. Yin, et al., Sci. Adv. 4 (2018) eaat5383. doi: 10.1126/sciadv.aat5383
S.S. Hwang, C.G. Cho, H. Kim, Electrochem. Commun. 12 (2010) 916–919. doi: 10.1016/j.elecom.2010.04.020
Q. Zhao, X. Liu, S. Stalin, K. Khan, L.A. Archer, Nat. Energy 4 (2019) 365–373. doi: 10.1038/s41560-019-0349-7
J.R. Nair, I. Shaji, N. Ehteshami, et al., M. Winter, Chem. Mater. 31 (2019) 3118–3133. doi: 10.1021/acs.chemmater.8b04172
D. Chen, M. Zhu, P. Kang, et al., Adv. Sci. 9 (2022) 2103663. doi: 10.1002/advs.202103663
J. Xiang, Y. Zhang, B. Zhang, et al., Energy Environ. Sci. 14 (2021) 3510–3521. doi: 10.1039/d1ee00049g
S. Wen, C. Luo, Q. Wang, et al., Energy Storage Mater. 47 (2022) 453–461. doi: 10.1016/j.ensm.2022.02.035
J. Zhu, J. Zhang, R. Zhao, et al., Energy Storage Mater. 57 (2023) 92–101. doi: 10.1016/j.ensm.2023.02.012
Z. Li, J. Fu, X. Zhou, et al., Adv. Sci. 10 (2023) 2201718. doi: 10.1002/advs.202201718
X. Liu, H. Jia, H. Li, Energy Storage Mater. 67 (2024) 103263. doi: 10.1016/j.ensm.2024.103263
Y. Seo, Y.C. Jung, M.S. Park, D.W. Kim, J. Membr. Sci. 603 (2020) 117995. doi: 10.1016/j.memsci.2020.117995
A. Mahmood, J. Energy Chem. 24 (2015) 686–692. doi: 10.1016/j.jechem.2015.10.018
X. Liu, Z. Xiao, H. Peng, et al., Chem. Asian J. 17 (2022) e202200929. doi: 10.1002/asia.202200929
N. Zhao, Y. Zou, X. Chen, et al., Colloid Surf. Physicochem. Eng. Asp. 666 (2023) 131317. doi: 10.1016/j.colsurfa.2023.131317
Q. Zhang, K. Liu, F. Ding, X. Liu, Nano Res. 10 (2017) 4139–4174. doi: 10.1007/s12274-017-1763-4
D. Zhang, L. Li, X. Wu, et al., Front. Energy Res. 9 (2021) 726738. doi: 10.3389/fenrg.2021.726738
T. Uno, H. Sano, M. Matsumoto, M. Kubo, T. Itoh, J. Solid State Electrochem. 14 (2010) 2161–2167. doi: 10.1007/s10008-009-0999-7
M. Jia, T. Li, D. Yang, et al., Batteries 9 (2023) 488. doi: 10.3390/batteries9100488
A.E. Abdelmaoula, L. Du, L. Xu, et al., Sci. China Mater. 65 (2022) 1476–1484. doi: 10.1007/s40843-021-1940-2
Z. Xiao, T. Long, L. Song, Y. Zheng, C. Wang, Ionics 28 (2022) 15–26. doi: 10.1007/s11581-021-04340-2
J. Chattopadhyay, T.S. Pathak, D.M.F. Santos, Polymers 15 (2023) 3907. doi: 10.3390/polym15193907
Z. Zhu, M. Xue, X. Wu, et al., Chem. Eng. J. 514 (2025) 163402. doi: 10.1016/j.cej.2025.163402
G. Xiao, K. Yang, Y. Qiu, et al., Adv. Mater. 37 (2025) 2415411. doi: 10.1002/adma.202415411
H. Xu, J. Mi, J. Ma, et al., Energy Environ. Sci. 18 (2025) 4231–4240. doi: 10.1039/d4ee05606j
Y. Yang, J. Li, S. Guo, et al., J. Colloid. Interface Sci. 699 (2025) 138306. doi: 10.1016/j.jcis.2025.138306
C. Wang, Z. Xia, G. Bai, et al., J. Energy Storage 132 (2025) 117730. doi: 10.1016/j.est.2025.117730

Figure 2 (a) General pathway for the preparation of SIPE films. Copied with permission [55]. Copyright 2016, American Chemical Society. (b) Schematics of synthesis of the P(SSPSILi-alt-MA) single-ion conductor. Copied with permission [56]. Copyright 2017, the Royal Society of Chemistry. (c) Schematic illustration of the azoles structures and the dissolution in the electrolyte. Copied with permission [57]. Copyright 2020, Elsevier Ltd. (d) Schematic diagram of the preparation process and the characteristics of FRSE. Copied with permission [58]. Copyright 2021, Elsevier B.V. (e) Schematic illustration of the in-situ preparation process (PTLiN1 sample). Copied with permission [59]. Copyright 2022, Elsevier B.V. (f) Schematic illustration of the ion-blocking protective layer's formation. Copied with permission [60]. Copyright 2022, The author(s). (g) Schematic illustration of the ion-blocking protective layer's formation. Copied with permission [61]. Copyright 2022, The author(s).

Figure 3 (a) Detailed synthetic scheme of 21-β-CD-g-PTFEMA. Copied with permission [68]. Copyright 2022, The author(s). (b) The polymerization process of TPDOL electrolyte. Copied with permission [69]. Copyright 2024, Wiley-VCH GmbH. (c) The mechanism of Li+ conduction in S-SPE and P-SPE. Copied with permission [70]. Copyright 2021, Published by Elsevier B.V. (d) Schematic illustration showing the chemical synthesis process of the electronegative PPFPA polymer brushes on the Celgard separator. Copied with permission [71]. Copyright 2022, Wiley-VCH GmbH. (e) Covalently grafted PVDF-CTFE-g-PEGA-co-PAA binder by ATRP. Copied with permission [72]. Copyright 2023, The authors. (f) Fabrication process of gel polymer electrolytes (PETEA-TCGG-PAN). Copied with permission [73]. Copyright 2022, American Chemical Society.

Figure 4 (a) Schematic illustrations of dual role for PPS polymer as artificial SEI in stabilizing Li electrode and solid polymer electrolyte for safe all-solid-state batteries. Copied with permission [74]. Copyright 2022, Elsevier B.V. (b) One-step in situ fabrication of block copolymer electrolytes. Copied with permission [75]. Copyright 2022, Wiley-VCH GmbH. (c) Schematic illustration of rationally fabricating hybrid copolymer electrolytes. Copied with permission [76]. Copyright 2024, American Chemical Society. (d) Schematic representation of structural modifications in the SEI induced by PPP. Copied with permission [79]. Copyright 2024, Elsevier B.V. (e) Schematic illustration of the design strategy of the branched PAA binder. Copied with permission [84]. Copyright 2019, WILEY‐VCH Verlag GmbH & Co. KGaA, Weinheim.

Figure 5 (a) Electrochemical redox reactions of a nitroxide radical. Copied with permission [85]. Copyright 2021, The Royal Society of Chemistry. (b) Schematic representation of the action of conjugated nitroxide radical polymer. Copied with permission [86]. Copyright 2024, American Chemical Society.

Figure 6 (a) Anionic polymerization mechanism initiated by metal Li. Copied with permission [104]. Copyright 2017, American Chemical Society. (b) Illustration of the Li+-transport mode in conventional SPE, pristine LiCOF, and DLC. Copied with permission [105]. Copyright 2022, Wiley-VCH GmbH. (c) Li metal battery enabling dendrite-free Li cycling at room temperature and thermal shutdown when temperatures increase. Copied with permission [106]. Copyright 2019, The Royal Society of Chemistry. (d) Schematic model of in situ generated polymer electrolyte via AROP. Copied with permission [107]. Copyright 2021, The Royal Society of Chemistry.

Figure 7 (a) Schematic model of the polymerization mechanism of DOL induced by LiPF6. Copied with permission [108]. Copyright 2018, The authors. (b) Schematic diagram of cationic polymerization of divinyl ethers. Copied with permission [109]. Copyright 2010, Elsevier B.V. (c) Schematic illustrating ex situ and in situ synthesis of SPEs. Copied with permission [110]. Copyright 2019, The author(s). (d) Schematic diagram of solvent-free polymerization. Copied with permission [111]. Copyright 2019, American Chemical Society. (e) Schematic of the preparation of self-enhancing gel polymer electrolyte. Copied with permission [112]. Copyright 2021, The authors. (f) Illustration of the integrated flexible all-solid-state lithium batteries. Copied with permission [114]. Copyright 2022, Elsevier B.V. (g) Schematic illustration of the in situ fabrication process of c-GPE and the crosslinking polymerization mechanism. Copied with permission [115]. Copyright 2023, Elsevier B.V.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:
 下载:
下载: