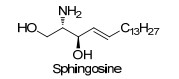
Figure 1.
Structure of sphinogosine
Synthesis of Azidosphingosine from D-Galactose or L-Arabinose
Yangguang Gao , Zhou Cao , Zhongxiang Han , Qiang Zhang , Jie Hu , Rui Guo , Xianran He , Fei Ding , Qingliang You , Yongmin Zhang

Sphingosine, named [(2S, 3R, 4E)-2-amino-3-hydroxyo- ctadec-4-en-1-ol)] (Figure 1), which is the active component of many sphingolipids, was firstly isolated from human brain by Thudichum in 1884.[1, 2] While sphingolipids are ubiquitously found in cell membrane of eukaryotic cells, and serve as versatile physiological functions.[3~6] Furthermore, sphingosine itself displays powerful inhibition of protein kinase C and plays critical role in signal pathway.[7, 8] Since the significance of sphingosine and its deficiency in nature, synthetic chemistry community has made a great endeavor to efficiently prepare optically pure sphingosine and its derivatives.
The main challenges towards synthesis of sphingosine have two points: one is to exquisitely set stereogenic centers at C2 and C3, another is to selectively construct E carbon-carbon double bond. A great number of papers about synthesis of sphingosine have been published in the past decades.[9~13] A typical synthesis of sphingosine was achieved by Zimmermann and Schmidt in 1988.[14] The highlight of their scheme was that D-galactose and D-xy- lose were respectively used as chiral precursor to provide 2, 4-O-protected D-threoses, upon which was treatment of ylide salt in the presence of an excess of lithium bromide to exclusively yield E-enetriols. More recently, Kitamura et al.[15] described a novel methodology on construction of 1, 2-amino alcohol via a catalytic asymmetric dehydrative allylation, which was applied to synthesize sphingosine within only five steps.
In a continuation of our review in 2016, [13] we have been working to develop a concise and satisfactory protocol to synthesize sphingosine and its derivatives. Azidosphingosine is not only a stable and important precursor of sphingosine, but also an excellent acceptor of glycosylation.[16] Consequently, synthesis of azidosphingosine counts for much. Herein, we described a modified synthesis of azidosphingosine from D-galactose. Besides, we developed a novel protocol for the synthesis of azidosphingosine from L-arabinose. Though D-arabinose has been used as chiral pool to synthesize D-erythro-sphingosine in the previous literature, [17] in which E-selective Wittig olefination was employed as key step. To the best of our knowledge, this is the first report about the synthesis of azidosphingosine from L-arabinose.
Synthesis of azidosphingosine (7) was summarized in Scheme 1. As shown in Scheme 1, firstly, D-galactose was protected as benzylidene ether at C4 and C6 with benzaldehyde dimethyl acetal in the presence of p-toluenesulfo- nic acid (PTSA) to provide compound 1 in a good yield (85%). Subsequent oxidative cleavage by sodium periodate in methanol was smoothly carried out to afford 2, 4-benzy- lidene-D-threose (2), which was directly subjected to the following Wittig olefination using the in situ generated methyl Wittig reagent to give compound 3 in 81% yield for 2 steps. Then, elongation of compound 3 was successfully achieved by olefin cross-metathesis (OCM) between compound 3 and 1-pentadecene (3a) under the catalysis of Grubbs II catalyst in dichloromethane (DCM) at reflux temperature to afford compound 4a in a yield of 62%, and albeit with 10% yield of compound 4b. To note, conversion of compound 2 into compound 4a underwent 2 steps other than one step in the previous literature, but the advantage of 2 steps operation was that it was unnecessary to use excess strong base such as PhLi[18] or BuLi-LiBr.[14] The undesired compound 4b could be converted into the E-olefin 4b by radical-induced isomerization[19] in a moderate yield (64%). Furthermore, when we repeated the same Wittig olefination according to the literature, no exclusive E-selectivity was observed, and the best result of the mixture of E, Z-alkenes in a ratio of 10:1 was given. Similar report can also be found in literature.[20] According to a literature procedure, [14, 18] compound 4a was initially converted to its triflate followed by substitution with NaN3 in dimenthylformamide (DMF). To our surprise, beside the desired azido product 5, 38% yield of elimination product compound 6 was also obtained. The possible reason for formation of elimination product can be explained as following: The triflate of compound 4a may underwent E1 elimination in polar solvent (DMF) in the presence of weak base (azide anion) to give vinyl benzylidene ether, and one equivalent of triflic acid was released. At the same time, vinyl benzylidene ether was unstable under acidic condition. Thus, the benzylidene group was removed to give enol compound, which then tautomerize to its keto form, i.e. compound 6. Because OTf usually works as a good leaving group, a less active group OMs was utilized instead. Unfortunately, subsequent replacement by NaN3 proved to be problematic, which only gave 40% yield of compound 5 for 2 steps. That was in good accordance with the previous result.[21] We were pleased to find when compound 4a was conducted under Mitsunobu’s condition, [22] the above problems such as elimination or low yield could be circumvented, and compound 5 was obtained with 72% yield. Treatment of compound 5 with AcCl in methanol provided azidosphingosine (7) in 89% yield. The overall yield of 27% over 6 steps is superior to 7% overall yield over 6 steps in the previous literature.[14]
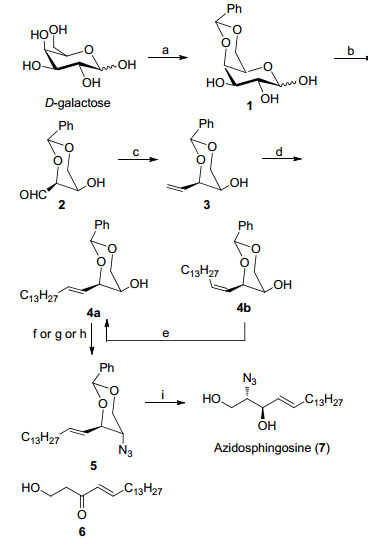
Reagents and conditions: (a) PhCH(OMe)2, PTSA, DMF (dry), 85%; (b) NaIO4, MeOH, H2O; (c) CH3Ph3P+·Br-, n-BuLi, THF, 81% for 2 steps; (d) 1-pentadecene (3a), Grubbs Ⅱ catalyst, 62% for 4a, 10% for 4b; (e) p-toluenethiol, AIBN, toluene, reflux, 64%; (f) Ph3P, DEAD, DPPA, THF (dry), 72%; (g) (ⅰ) Tf2O, DCM, pyridine (dry), -20 ˚C to r.t.; (ⅱ) NaN3, DMF (dry), 45% for 5, 38% for 6; (h) (ⅰ) MsCl, DCM, Et3N, -30 ˚C to r.t.; (ⅱ) NaN3, DMF, 90 ˚C, 40%; (ⅰ) 1% AcCl in methanolic solution, 89%.
The second-generation synthesis of azidosphingosine (7) was depicted in Scheme 2. L-Arabinose was converted into compound 8 according to the previously reported procedure.[19] Unblocking of compound 8 in acidic solution followed by regioselective protection as benzylidene ether provided compound 9 in 85% yield for 2 steps. Initially, compound 9 was subjected to olefin cross-metathesis (OCM) with 1-pentadecene (3a) under the catalysis of Grubbs II catalyst. To our surprise, poor E-selectivity in CM reaction was observed, and the mixture of Z, E-alkenes was inseparable by routine column chromatography. The- refore, as a candidate route, Appel reaction[23] of compound 9 was carried out with I2, PPh3, imidazole in tetra- hydrofuran (THF) to give a 91% yield of compound 11. Unfortunately, subsequent azido replacement of iodo product 11 by NaN3 proved to be unsuccessful. Elimination product was obtained unexpectedly. To overcome the problem, we envisaged that elongation of the double bond might suppress the elimination due to the increasing electronic effect of the olefin. Thus, olefin cross-metathesis (OCM) between compound 11 and 1-pentadecene (3a) was executed to furnish compound 13 in 90% yield. To note, better E-selectivity (E/Z=10/1) was observed in OCM reaction than its counterpart compound 3 or 9. Besides, compound 13 can be more easily separated from its cis byproduct. However, the following replacement by NaN3 in DMF failed to provide compound 14, and the elimination product compound 6 was still obtained instead. At this stage, we began to doubt that the presence of benzylidene group at C1 and C3 may promote the elimination of hydrogen at allylic position due to its conjugated effect. As expected, removal of benzylidene group followed by azido substitution gave azidosphingosine (7) successfully in a yield of 82%. The overall yield was 20% over 8 independent manipulations. The spectral and analytical data (1H NMR, 13C NMR, HRMS and optical rotation values) of synthetic azidosphingosine were in good accordance with the previously reported literatures.
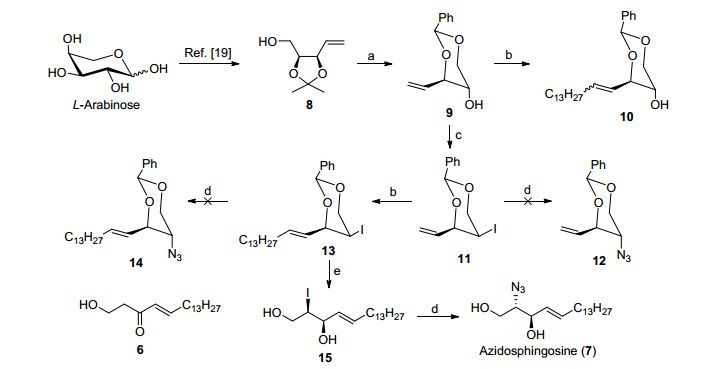
Reagents and conditions: (a) (ⅰ) PTSA, MeOH; (ⅱ) PhCH(OMe)2, CH3CN, 85% yield for 2 steps; (b) 1-pentadecene, Grubbs Ⅱ catalyst, 65% yield for 10, 90% yield for 13; (c) Ⅰ2, PPh3, imidazole, THF, 91% yield; (d) NaN3, DMF, 50 ℃, 75% yield for 7; (e) PTSA, MeOH, 82% yield.
In summary, a modified synthesis of azidosphingosine from D-galactose has been achieved, besides, a novel synthesis of azidosphingosine from L-arabinose has been developed. The overall yields were 27% and 20%, respectively, and both were better than that of the previous literature. Both routes underwent a similar benzylidene-pro- tected enetriol, and OCM reaction was utilized as key step to elong the carbon-carbon double bond. The disfavored elimination during azido replacement was reported for the first time, and the reason for formation of elimination product was also presumed. Our protocol is flexible and effective, which could provide alternative syntheses of sphingosine and its diastereoisomers using chiral template. And these results will be reported in due course.
All reagents were commercially available and used directly without further purification unless otherwise stated. Column chromatography was carried out by using silica gel (100~200 mesh). Routine monitoring of reactions was used silica gel60 F254 TLC plates. Dried THF was distilled from Na and benzophenone. DMF, DCM were treated with CaH2. All air-sensitive reactions were run under a nitrogen atmosphere. Purification by column chromato- graphy was using 100~200 mesh silica gel. Reactions were monitored by TLC using silica gel 60 F254 plates. 1H NMR and 13C NMR spectra were recorded with a Bruker Avance III spectrometer at 400 and 100 MHz respectively relative to Me4Si (δ=0) as internal standard. HRMS were measured with a ThermoFisher Q-Exactive mass spec-trometer. IR spectra were recorded on neat sample with a NICOLET iS 10 infrared spectrometer. Optical rotations were recorded with a AUTOPOL IV automatic polarimeter.
D-Galactose (9.00 g, 0.05 mol) was dissolved in 100 mL of dried N, N-dimethyl formide (DMF). To the resulting solution was added benzaldehyde dimethyl acetal (12 mL, 0.08 mol), p-toluenesulfonic acid (PTSA) (1.00 g, 5.80 mmol) in sequence. The mixture was allowed to be heated at 50 ℃, then 1 mL of Et3N was added to quench the reaction until the starting material was throughly consumed (monitored by TLC). The solution was concentrated in vacuo and column chromatography of the residue on silica gel (EtOAc-MeOH, V:V=10:1) afforded a mixture of α/β-1 (α/β=5/1 analyzed by 1H NMR) as a white solid (11.39 g, 85%). m.p. 180~183 ℃ (Lit.[18] m.p. 171~173 ℃);
Compound 1 (2.00 g, 7.46 mmol) was completely dissolved in 30 mL of methanol under sonication condition, then the solution was placed on an ice bath. To the solution was added sodium periodate (2.40 g, 11.22 mmol) in water (30 mL) via dropping funnel. The resulting solution was stirred at ambient temperature till the starting material throughly disappeared. The undissolved white solid was filtered, and the filtrate was extracted with DCM (50 mL×3), combined the organic phase, concentrated, and the yellow residue was directly subjected to the next step without further purification.
To a suspension of methyltriphenylphosphonium bro- mide (5.00 g, 14.00 mmol) in anhydrous THF (30 mL) was added n-BuLi solution (7.8 mL, 2.2 mol·L-1 in n-hexane, 17.16 mmol) dropwise at 0 ℃ under a nitrogen atmosphere. The mixture was turned to yellow immediately, 45 min later, to the yellow suspension was gradually added a solution of crude compound 2 (2.00 g, 9.60 mmol) in 10 mL of THF. Saturated ammonia chloride solution (20 mL) was added to quench the reaction until the completion of the reaction, then the organic layer was separated, and extracted with EtOAc (25 mL×3), dried over anhydrous Na2SO4, and the organic layer was concentrated. The residue was purified by silica gel column chromatography (petroleum ether- EtOAc, V:V=6:1) to furnish compound 3 (1.60 g, 81% for 2 steps) as a white powder. m.p. 40~42 ℃;
To a solution of 1-bromopentadecane (1.00 g, 3.43 mmol) in 10 mL of THF (dried) was added t-BuOK (1.12 g, 10.00 mmol), the solution was stirred at room temperature until the reaction completed (monitored by TLC). Saturated ammonia chloride solution was poured into the above solution to quench the reaction, then extracted with EtOAc (20 mL×3), dried over anhydrous Na2SO4, and concentrated under reduced pressure. Column chromatography (pure petroleum ether) of the residue gave compound 3a as a colorless liquid (0.72 g, 99%). 1H NMR (CDCl3, 400 MHz) δ: 5.87~5.77 (m, 1H), 5.99 (dd, J=17.2, 2.0 Hz, 1H), 4.93 (dd, J=10.4, 0.8 Hz, 1H), 2.04 (q, J=7.2 Hz, 2H), 1.40~1.34 (m, 2H), 1.40~1.31 (m, 20H), 0.89 (t, J=7.2 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ: 139.4, 114.1, 33.9, 32.0, 29.8, 29.6, 29.4, 29.2, 29.0, 22.8, 14.2; IR (neat) ν: 3077, 2950, 1641, 1470, 1361 cm-1; HRMS (ESI, positive) calcd for C15H30Na [M+Na]+ 233.2245, found 233.2239.
To the mixture of compound 3 (600 mg, 2.91 mmol) and 3a (920 mg, 4.38 mmol) in dried DCM (10 mL) under a nitrogen atmosphere was added a catalyzed amount of Grubbs II catalyst (123 mg, 0.15 mmol). The solution was allowed to be heated at reflux temperature for 8 h, then concentrated under reduced pressure. The residue was purified by column chromatography (toluene-EtOAc, V:V=50:1) to give compound 4a as a white solid (701 mg, 62%), and compound 4b as a colorless liquid (113 mg, 10%). For 4a: m.p. 52~54 ℃ (Lit.[18] m.p. 55~56 ℃);
To a solution of compound 4b (100 mg, 0.26 mmol) in anhydrous toluene (8 mL) was added p-toluenethiol (320 mg, 2.58 mmol), AIBN (424 mg, 2.58 mmol) under a nitrogen atmosphere. The mixture was heated at reflux for 8h, then concentrated in vacuo. The residue was purified by column chromatography (toluene-EtOAc, V:V=50:1) to give compound 4a (64 mg, 64%), and recovered 22 mg of compound 4b.
Method A: To a solution of compound 4a (1.00 g, 2.58 mmol) in 10 mL of dried THF under a nitrogen atmosphere was added PPh3 (2.70 g, 10.30 mmol), diethyl azodiformate (DEAD) (1.55 mL, 10.3 mmol), diphenyl azidophosphate (DPPA) (0.22 mL, 10.3 mmol) in sequence. The mixture was stirred at room temperature until the completion of the reaction, concentrated. The residue was purified by column chromatography (petroleum ether-EtOAc, V:V=50:1) to give compound 5 as a white solid (766 mg, 72%). m.p. 41~43 ℃ (Lit.[18] m.p. 43 ℃);
Method B: To a solution of compound 4a (150 mg, 0.39 mmol) in 6 mL of dried DCM was added 0.6 mL of dried pyridine and Tf2O (0.15 mL, 0.89 mmol) at -20 ℃ under a nitrogen atmosphere. After the disappearance of the starting material, the solution was concentrated in vacuo. NaN3 (140 mg, 2.15 mmol) was added to the above residue followed by addition of DMF (3 mL). The mixture was allowed to stir at room temperature until the reaction completed, filtered the solid, the filtrate was concentrated in vacuo. Column chromatography (petroleum ether-EtOAc, V:V=50:1) of the residue provided compound 5 as a white powder (71.8 mg, 45%), compound 6 as a colorless liquid (41 mg, 38%). For 6: 1H NMR (CDCl3, 400 MHz) δ: 6.88 (dt, J=15.6, 14.8 Hz, 1H), 6.10 (d, J=16.0 Hz, 1H), 3.89 (t, J=4.8 Hz, 2H), 2.81 (t, J=5.2 Hz, 2H), 2.22 (q, J=7.2 Hz, 2H), 1.48~1.44 (m, 2H), 1.25 (m, 20H), 0.88 (t, J=6.4 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ: 201.0, 149.2, 130.5, 58.2, 41.3, 32.6, 32.0, 29.8, 29.7, 29.7, 29.6, 29.5, 29.4, 29.3, 28.1, 22.8, 14.2; HRMS (ESI, positive) calcd for C18H34O2Na [M+Na]+ 305.2457, found 305.2478..
Method C: To a solution of compound 4a (350 mg, 0.90 mmol) in 8 mL of dried DCM was added 0.62 mL of Et3N and MsCl (0.16 mL, 1.80 mmol) at -30 ℃ under a nitrogen atmosphere. After the disappearance of the starting material, the solution was concentrated under reduced pressure. The resulting white powder was dissolved in 10 mL of dried DMF, then NaN3 (292 mg, 4.50 mmol) was added. The suspension was heated at 90 ℃ for 3 d till the starting material was consumed. 10 mL of water was poured into the suspension, extracted with petroleum ether, combined the organic layer, and evaporated. The residue was purified by column chromatography (petroleum ether-EtOAc, V:V=50:1) to furnish compound 5 (149 mg, 40%).
Method A: To a solution of compound 5 (800 mg, 1.93 mmol) in methanol (4 mL) was added AcCl (40 μL). The solution was allowed to stir at room temperature until the consumption of the starting material. The reaction was quenched by addition of 0.1 mL of Et3N, then concentrated. Column chromatography (petroleum ether-EtOAc, V:V=2:1) of the residue provided compound 7 as a white solid (560 mg, 89%). m.p. 52~54 ℃ (Lit.[18] m.p. 52~54 ℃);
Method B: To a solution of compound 15 (100 mg, 0.24 mmol) in DMF (3 mL) was added NaN3 (159 mg, 2.44 mmol). The mixture was heated at 50 ℃ till the starting material was throughly consumed. The solvent was concentrated in vacuo, the residue was added 10 mL of DCM, filtered. The filtrate was concentrated. Column chromatogrphy (petroleum ether-EtOAc, V:V=2:1) of the residue provided compound 7 as a white solid (59 mg, 75%).
To a solution of compound 8 (500 mg, 3.16 mmol) in 10 mL of MeOH was added PTSA (60 mg, 0.32 mmol). The mixture was allowed to stir at room temperature, and the solvent was removed under reduced pressure till the completion of the reaction. The residue was dissolved in 10 mL of CH3CN, then, benzaldehyde dimethyl acetal (0.6 mL, 4.00 mmol) was added. 2 mL of Et3N was added to quench the reaction until the starting material completely disappeared (monitored by TLC). The mixture was concentrated, column chromatography of the residue (petroleum ether-EtOAc, V:V=50:1) gave compound 9 as a white solid (554 mg, 85%). m.p. 82~85 ℃;
To a solution of compound 9 (700 mg, 3.39 mmol) in 20 mL of THF (dried) was added I2 (1.03 g, 4.06 mmol), PPh3 (1.34 g, 5.10 mmol), imidazole (0.69 g, 10.13 mmol) in sequence. The mixture was heated at reflux temperature for 5 h. Saturated Na2S2O3 solution was added to quench the reaction, extracted with EtOAc (20 mL×3), dried over anhydrous Na2SO4 and concentrated. The residue was purified by column chromatography (petroleum ether-EtOAc, V:V=6:1) to afford compound 11 as a white powder (976 mg, 91%). m.p. 66~73 ˚C;
To a mixture of compound 11 (600 mg, 1.90 mmol) and 1-pentadecene (3a) (800 mg, 3.80 mmol) was added Grubbs II catalyst (81 mg, 0.09 mmol). The solution was heated at reflux temperature. The solvent was removed until the starting material was throughly consumed. Column chromatography (petroleum ether-EtOAc, V:V= 10:1) of the residue provided compound 13 as a white powder (852 mg, 90%). m.p. 52~57 ℃;
Compound 13 (500 mg, 1.00 mmol) was dissolved in 10 mL of MeOH. To the solution was added PTSA (10 mg, 0.05 mmol). 0.1 mL of Et3N was added to neutralize the acid after the completion of the reaction, then the solvent was removed in vacuo. The residue was purified by column chromatography (petroleum ether-EtOAc, V:V=2:1) to afford compound 15 as an amorphous powder (337 mg, 82%).
Supporting Information 1H NMR and 13C NMR spectra for compounds 1, 3, 3a, 4a, 4b, 5~7, 9, 11, 13 and 15. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn/.
Thudichum, J. L. W. A Treatise on the Chemical Constitution of the Brain, Bailliere, Tindall, and Cox, London, 1884. http://www.jstor.org/stable/228216
Du, H. W.; Chen, J. X.; Hao, J. S.; Pan, J. G.; Xia, C. Z. Chem. J. Chin. Unive. 1999, 20, 590.
Chang, Y. T.; Choi, J.; Ding, S.; Prieschl, E. E.; Baumruker, T.; Lee, J. M.; Chung, S. K.; Schultz, P. G. J. Am. Chem. Soc. 2002, 124, 1856. doi: 10.1021/ja017576o
Merrill, A. H. Jr.; Sandhoff, K. In Biochemistry of Lipids, Lipoproteins and Membranes, Eds.:Vance, D. E.; Vance, J. E., Elsevier, New York, 2002, p. 373.
Lee, J. Y.; Hannun, Y. A.; Obeid, L. M. J. Biol. Chem.1996, 271, 13169. doi: 10.1074/jbc.271.22.13169
Hannun, Y. A. J. Biol. Chem. 1994, 269, 3125. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM3101176
Hannun, Y. A.; Bell, R. M. Science 1989, 243, 500. doi: 10.1126/science.2643164
Merill, A. H. J. Bioenerg. Biomembr. 1991, 23, 83.
Howell, A. R.; Ndakala, A. J. Curr. Org. Chem. 2002, 6, 365. doi: 10.2174/1385272024604998
Koskinen, P. M.; Koskinen, A. M. P. Synthesis 1998, 1075.
Morales-Serna, J. A.; Llaveria, J.; Díaz, Y.; Matheu, M. I.; Cas-tillón, S. Curr. Org. Chem. 2010, 14, 2483. doi: 10.2174/138527210793358286
Liao, J. Y.; Tao, J. H.; Lin, G. Q.; Liu, D. G. Tetrahedron 2005, 61, 4715. doi: 10.1016/j.tet.2005.02.075
Gao, Y. G.; He, X. R.; Ding, F.; Zhang, Y. M. Synthesis 2016, 48, 4017. doi: 10.1055/s-0036-1588311
Zimmermann, P.; Schmidt, R. R. Liebigs Ann. Chem. 1988, 663. http://www.researchgate.net/publication/243929006_Synthese_vonerythro-Sphingosinen_ber_die_Azidoderivate
Vankar, Y. D.; Schmidt, R. R. Chem. Soc. Rev. 2000, 29, 201. doi: 10.1039/a900943d
Schmidt, R. R.; Zimmermann, P. Tetrahedron Lett. 1986, 27, 481. doi: 10.1016/S0040-4039(00)85510-0
Tanaka, S.; Gunasekar, R.; Tanaka, T.; Iyoda, Y.; Suzuki, Y.; Kitamura, M. J. Org. Chem. 2017, 82, 9160. doi: 10.1021/acs.joc.7b01181
Duclos Jr, R. I. Chem. Phys. Lipids 2001, 111, 111. doi: 10.1016/S0009-3084(01)00152-9
Gao, Y. G.; Cao, Z.; Su, C. Z.; Chen, Z. F.; He, X. R.; Ding, F.; Li, H.; Zhang, Y. M. Synthesis 2016, 48, 4471. doi: 10.1055/s-0036-1588343
Ohashi, K.; Yamagiwa, Y.; Kamikawa, T.; Kates, M. Tetrahedron Lett. 1988, 29, 1185. doi: 10.1016/S0040-4039(00)86683-6
Compostella, F.; Franchini, L.; Panza, L.; Prosperi, D.; Ronchetti, F. Tetrahedron 2002, 58, 4425. doi: 10.1016/S0040-4020(02)00418-0
(a) Niu, Y.; Cao, X.; Ye, X. S. Helv. Chim. Acta 2008, 91, 746.
(b) Jacqueline, E. M.; Krzysztof, J.; Philip, J. K.; Jorge, A. Chem. Commun. 2002, 426.
(a) Appel, R. Angew. Chem. 1975, 87, 863.
(b) Denton, R. M.; An, J.; Adeniran, B.; Blake, A. J.; Lewis, W.; Poulton, A. M. J. Org. Chem. 2011, 76, 6749.

Scheme 1 Synthesis of azidosphingosine (7)
Reagents and conditions: (a) PhCH(OMe)2, PTSA, DMF (dry), 85%; (b) NaIO4, MeOH, H2O; (c) CH3Ph3P+·Br-, n-BuLi, THF, 81% for 2 steps; (d) 1-pentadecene (3a), Grubbs Ⅱ catalyst, 62% for 4a, 10% for 4b; (e) p-toluenethiol, AIBN, toluene, reflux, 64%; (f) Ph3P, DEAD, DPPA, THF (dry), 72%; (g) (ⅰ) Tf2O, DCM, pyridine (dry), -20 ˚C to r.t.; (ⅱ) NaN3, DMF (dry), 45% for 5, 38% for 6; (h) (ⅰ) MsCl, DCM, Et3N, -30 ˚C to r.t.; (ⅱ) NaN3, DMF, 90 ˚C, 40%; (ⅰ) 1% AcCl in methanolic solution, 89%.

Scheme 2 Second-generation synthesis of azidosphingosine (7)
Reagents and conditions: (a) (ⅰ) PTSA, MeOH; (ⅱ) PhCH(OMe)2, CH3CN, 85% yield for 2 steps; (b) 1-pentadecene, Grubbs Ⅱ catalyst, 65% yield for 10, 90% yield for 13; (c) Ⅰ2, PPh3, imidazole, THF, 91% yield; (d) NaN3, DMF, 50 ℃, 75% yield for 7; (e) PTSA, MeOH, 82% yield.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:
 下载:
下载: