
Scheme 1.
Radical decarboxylative borylation of RAEs.
Electrochemically promoted decarboxylative borylation of alkyl N-hydroxyphthalimide esters
Jian-Jun Dai , Xin-Xin Teng , Wen Fang , Jun Xu , Hua-Jian Xu

Alkyl boronic acid and esters represent important scaffolds in both organic synthesis and pharmaceuticals [1]. For example, alkyl boronic acid and esters show great value for the construction of C(sp3)–rich scaffolds using cross-coupling reactions [2, 3]. In addition, molecules containing alkyl boron structures have been proven to be life-saving drugs such as Velcade [4, 5]. Classic methods to synthesize alkyl boron compounds include hydroboration of alkenes [6-11], borylation of organometallic nucleophiles (e.g., Grignard and organolithium reagents) [12-14]. However, these methods suffer from harsh reaction conditions and poor functional group tolerance. Over the past decade, great attention has been directed to the borylation of alkyl halides using transition-metal catalysis including Cu [15, 16], Ni [17, 18], Pd [17, 19], Zn [20], Fe [21, 22], Mn [23] and more recently photo-induced catalysis [24-33]. In recent years, the use of easily available and stable carboxylic acids redox active esters (RAEs, e.g., N-hydroxyphthalimide esters) derived from alkyl carboxylic acids as starting materials for decarboxylative transformations is a fast growing research area [34-40]. In particular, remarkable progresses have been made with regard to decarboxylative borylation of RAEs [41-44]. Baran and co-workers reported transition-metal catalyzed (i.e., nickel, copper) protocols for the decarboxylative borylation of RAEs with bis(pinacolato)diboron (B2pin2) using excess MeLi and LiOH as the base, respectively [45, 46]. Li and co-workers described the decarboxylative borylation of RAEs with excess B2pin2 using an Ir-based photoredox catalyst under 45 W CFL irradiation (Scheme 1A) [47]. Aggarwal et al. developed an efficient photo-induced method to achieved the decarboxylative borylation of RAEs with bis(catecholato)diboron (B2cat2) under irradiation with blue LEDs (Scheme 1A) [48]. An important feature to the success of these decarboxylative transformations of RAEs is the facile generation of alkyl radical intermediates via single-electron transfer process.
Electro-organic synthesis servers as a powerful and green technique for oxidative and reductive reactions by single-electron transfer under mild conditions and good functional group tolerance [49-56]. Recently, elegant works on electrochemical reductive decarboxylative carbon–carbon bond formation using RAEs have been disclosed by Wang [57], Lei [58] and Zeng [59] groups.
In this context, we hypothesized that the electrochemically promoted reductive decarboxylation of RAEs may be compatible with diboron reagents such as B2pin2, B2cat2, thus resulting the decarboxylative borylation reaction. Herein, we report an efficient method for the construction of alkyl boronic esters using an electrochemically promoted decarboxylative borylation of alkyl N-hydroxyphthalimide esters (Scheme 1B). This reaction exhibits broad substrate scope, good functional group tolerability, and easy scalability, representing an alternative method for the formation of C(sp3)–B bond with electrochemical techniques [60, 61].
We started our investigation with the electrochemical decarboxylative borylation of N-(cyclohexanecarbonyl)phthalimide and B2cat2 under constant current electrolysis (CCE) (Table 1). It was found that the desired product 1 can be obtained in 86% yield (76% isolated) with 10 mA constant current in an undivided cell followed by the treatment of pinacol and Et3N (entry 1). The use of other supporting electrolytes including nBu4NPF6, LiClO4 and NaBF4 (entries 2–4) gave 1 in slightly lower yields. Increasing or decreasing the electric current delivered diminished yields (entries 5 and 6). It was also found that this reaction was sensitive to air (entry 7). The cathode materials such as reticulated vitreous carbon (RVC), nickel, copper and iron failed to improve the efficiency of this reaction (entry 8, please see Supporting information for more details). Also, the use of graphite anode resulted in lower efficiency compared to that of platinum anode (entry 9). Notably, the solvent is important to the outcome of the reaction: the use of THF gave a decreased yield (entry 10), however the performances of other solvents such as MeCN, DMSO and NMP were poorer than that of DMF (entries 11–13). Additionally, the decarboxylative borylation reaction proceeded well at cathode in a divided cell (entry 14, not optimized). It should be noted that the reaction can be promoted by light from fume hood white bulb but at a significantly lower rate (entry 15). This result is in agreement with the previous report by Aggarwal and co-workers [48]. Finally, control experiments showed that a similar positive result was obtained when the reaction was conducted in dark (entry 16) and a very lower yield of 1 was observed without the use of electric current (entry 17). These results highlight the crucial role of an electric current in the decarboxylative borylation reaction.
With the optimal reaction conditions in hand, the scope of this reaction was then examined with respect to different alkyl carboxylic acids. As summarized in Scheme 2A, primary N-hydroxyphthalimide esters bearing either electron-donating or electron-withdrawing groups were smoothly converted to the corresponding alkylboronates in moderate to good yields (39%–62%). It was demonstrated that halides such as fluoro (14), chloro (3 and 15) and bromo (7 and 16) groups were well compatible with the reaction, thus not only demonstrating the orthogonal reactivity of this reaction with previously developed transition metal-catalyzed borylation of halides [15-23], but also providing opportunities for further conversions with cross-coupling reactions [3]. Furthermore, N-hydroxyphthalimide esters possessing terminal olefin (4), ester (5), phenyl (8 and 11), methyl (12), methoxy (9 and 13), thienyl (10) and trifluoromethyl (17) groups were well tolerated.
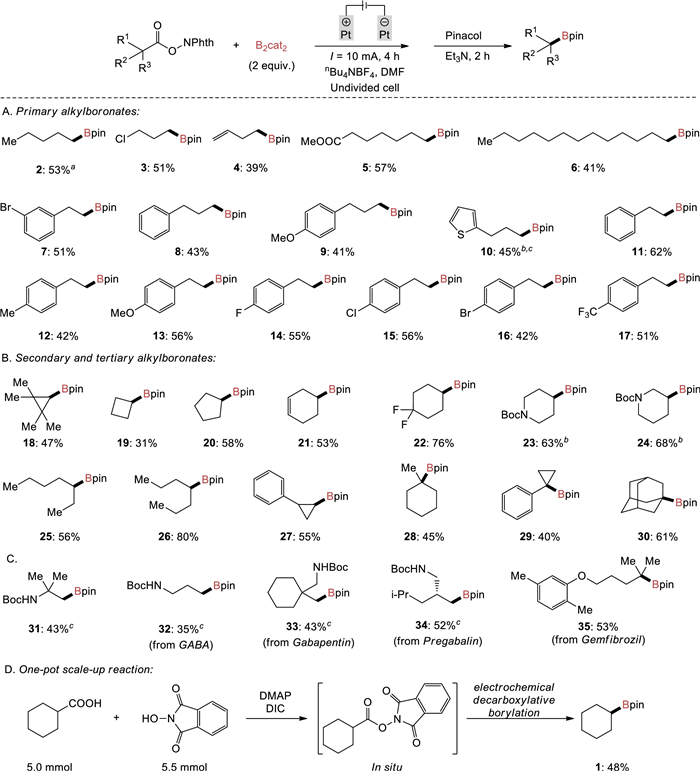
Then, investigation of the substrate scope was focused on secondary and tertiary N-hydroxyphthalimide esters (Scheme 2B). Both acyclic and cyclic alkyl N-hydroxyphthalimide esters were all suitable for the decarboxylative borylation, yielding the desired secondary and tertiary alkylboronates in moderate to good yields (31%–80%). For example, cyclohexyl (2, 22 and 28), cyclopentyl (20), inner olefin (21), cyclopropyl (18, 27 and 29) and adamantyl (30) motifs were tolerated, as was the important heterocycle, Boc-protected piperidine (23 and 24). The cyclobutyl boronic ester (19) could also be achieved, albeit with a lower yield. Interestingly, Aggarwal et al. reported tertiary carboxylic acids such as 1-methyl-cyclohexyl carboxylic acid failed to provide the desired decarboxylative borylation product in photochemical conditions [48]. To our delight, the desired boronic ester (28) can be formed smoothly under our electrochemical conditions, suggesting that this electrochemical protocol is complementary to that previously developed photochemical process.
To further expand the substrate scope, we conducted late-stage modifications of amino acids and drug molecules (Scheme 2C). Specifically, β-amino acid and γ-amino acids (e.g., γ-aminobutyric acid (GABA), gabapentin and pregabalin) were converted into boronic esters (31–34) in moderate yields. Gemfibrozil, a lipid-lowering drug, gave the corresponding boronic ester 35 in 53% yield. The reaction was also amenable to scale-up by a one-pot procedure in which cyclohexyl N-(cyclohexanecarbonyl)phthalimide formed in situ, followed by electrochemically promoted decarboxylative borylation, delivering the corresponding boronic ester 1 in 48% yield on a 5.0 mmol scale (Scheme 2D).
A series of experiments were conducted to gain more mechanistic insights of the electrochemically promoted decarboxylative borylation reaction (Scheme 3). In the radical trapping experiment, with the use of 2, 2, 6, 6-tetramethylpiperidine 1-oxyl (TEMPO, 3 equiv.) as a trapping agent under the standard conditions, the formation of 1 was obviously inhibited and the cyclohexyl/TEMPO adduct 37 was detected in HRMS analysis (Scheme 3A). Further evidence for the radical mechanism was provided by the reaction of 38, where only ring-opening product 4 was obtained without the formation of cyclopropyl-containing 39 (Scheme 3B). Moreover, when the 5-hexenyl radical clock precursor 40 was used, we obtained cyclization reaction product 41 in 17% yield along with linear product 42, the ratio of 41 and 42 was determined by 1H NMR analysis (Scheme 3B).

Based on previous reports of radical borylation [24, 30, 34] and the above experimental results, we proposed a possible mechanism for the electrochemically promoted decarboxylative borylation reaction (Scheme 3C). Initially, alkyl N-hydroxyphthalimide ester was reduced at the cathode to generate radical anion A that underwent decarboxylation to form the corresponding alkyl radical B intermediacy. Then, alkyl radical B reacts with B2cat2 and DMF to deliver the intermediate C. Subsequently, the cleavage of B–B bond of C to generate D and F. Finally, D is converted to product E with the treatment of pinacol/triethylamine. The radical F is eventually oxidized at the anode yielding G, and reaction of G and NPhth anion to deliver H.
In summary, we have developed an electrochemically promoted decarboxylative borylation reaction. The process shows broad substrate scope and provides a series of alkyl boronic esters. It is worth noting that the diboron reagents compatible well with electrochemical reaction conditions, which will provide opportunities for discovering new electrochemical borylation reactions. The use of electrochemical methods for making other type of organon boron compounds is currently underway in our laboratory.
We declare that we have no financial and personal relationships with other people or organizations that can inappropriately influence our work, there is no professional or other personal interest of any nature or kind in any product, service and/or company that could be construed as influencing the position presented in, or the review of, the manuscript entitled.
This work was supported by the National Natural Science Foundation of China (Nos. 21871074 and 21971051), the National Key R & D Program of China (No. 2018YFB1501604), the Fundamental Research Funds for the Central Universities (No. PA2020GDKC0021) and the Key Research and Development Program Projects in Anhui Province (No. 201904a07020069).
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.09.011.
D.G. Hall, Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials, Wiley-VCH, 2011.
N. Miyaura, A. Suzuki, Chem. Rev. 95 (1995) 2457–2483. doi: 10.1021/cr00039a007
A. de Meijere, F. Diederich, Metal-Catalyzed Cross-Coupling Reactions, 2nd ed., Wiley-VCH, Weinheim, 2004.
T. Hideshima, P. Richardson, D. Chauhan, et al., Cancer Res. 61 (2001) 3071–3076.
A. Paramore, S. Frantz, Nat. Rev. Drug Discovery2 (2003) 611–612. doi: 10.1038/nrd1159
G.C. Fu, Transition Metal-Catalyzed Hydroboration of Olefins. Transition Metals for Organic Synthesis: Building Blocks and Fine Chemicals, 2nd ed., Wiley-VCH, Weinheim, 2004.
K. Burgess, M.J. Ohlmeyer, Chem. Rev. 91 (1991) 1179–1191. doi: 10.1021/cr00006a003
K. Burgess, W.A. Van der Donk, S.A. Westcott, et al., J. Am. Chem. Soc. 114 (1992) 9350–9359. doi: 10.1021/ja00050a015
D.A. Evans, G.C. Fu, A.H. Hoveyda, J. Am. Chem. Soc. 114 (1992) 6671–6679. doi: 10.1021/ja00043a009
C.M. Crudden, D. Edwards, Eur. J. Org. Chem. 24 (2003) 4695–4712.
A.M. Carroll, T.P. O'Sullivan, P. J. Guiry, Adv. Synth. Catal. 347 (2005) 609–631. doi: 10.1002/adsc.200404232
E. Khotinsky, M. Melamed, Ber. Dtsch. Chem. Ges. 42 (1909) 3090–3096. doi: 10.1002/cber.19090420327
E. Krause, R. Nitsche, Ber. Dtsch. Chem. Ges. 54 (1921) 2784–2791. doi: 10.1002/cber.19210541027
H.C. Brown, T.E. Cole, Organometallics2 (1983) 1316–1319. doi: 10.1021/om50004a009
C.T. Yang, Z.Q. Zhang, H. Tajuddin, et al., Angew. Chem. Int. Ed. 51 (2012) 528–532. doi: 10.1002/anie.201106299
H. Ito, K. Kubota, Org. Lett. 14 (2012) 890–893. doi: 10.1021/ol203413w
J. Yi, J.H. Liu, J. Liang, et al., Adv. Synth. Catal. 354 (2012) 1685–1691. doi: 10.1002/adsc.201200136
A.S. Dudnik, G.C. Fu, J. Am. Chem. Soc. 134 (2012) 10693–10697. doi: 10.1021/ja304068t
A. Joshi-Pangu, X. Ma, M. Diane, et al., J. Org. Chem. 77 (2012) 6629–6633. doi: 10.1021/jo301156e
S.K. Bose, K. Fucke, L. Liu, P.G. Steel, T.B. Marder, Angew. Chem. Int. Ed. 53 (2014) 1799–1803. doi: 10.1002/anie.201308855
T.C. Atack, R.M. Lecker, S.P. Cook, J. Am. Chem. Soc. 136 (2014) 9521–9523. doi: 10.1021/ja505199u
R.B. Bedford, P.B. Brenner, E. Carter, et al., Organometallics33 (2014) 5940–5943. doi: 10.1021/om500847j
T.C. Atack, S.P. Cook, J. Am. Chem. Soc. 138 (2016) 6139–6142. doi: 10.1021/jacs.6b03157
Y. Cheng, C. Mück-Lichtenfeld, A. Studer, Angew. Chem. Int. Ed. 57 (2018) 16832–16836. doi: 10.1002/anie.201810782
D. Mazzarella, G. Magagnano, B. Schweitzer-Chaput, P. Melchiorre, ACS Catal. 9 (2019) 5876–5880. doi: 10.1021/acscatal.9b01482
L. Zhang, Z.Q. Wu, L. Jiao, Angew. Chem. Int. Ed. 59 (2020) 2095–2099. doi: 10.1002/anie.201912564
J. Wu, L. He, A. Noble, V.K. Aggarwal, J. Am. Chem. Soc. 140 (2018) 10700–10704. doi: 10.1021/jacs.8b07103
F. Sandfort, F. Strieth-Kalthoff, F.J.R. Klauck, M.J. James, F. Glorius, Chem. Eur. J. 24 (2018) 17210–17214. doi: 10.1002/chem.201804246
J. Hu, G. Wang, S. Li, Z. Shi, Angew. Chem. Int. Ed. 57 (2018) 15227–15231. doi: 10.1002/anie.201809608
F.W. Friese, A. Studer, Angew. Chem. Int. Ed. 58 (2019) 9561–9564. doi: 10.1002/anie.201904028
J. Wu, R.M. Bar, L. Guo, A. Noble, V.K. Aggarwal, Angew. Chem. Int. Ed. 58 (2019) 18830–18834. doi: 10.1002/anie.201910051
Q. Liu, J. Hong, B. Sun, et al., Org. Lett. 21 (2019) 6597–6602. doi: 10.1021/acs.orglett.9b01951
F. W. Friese, A. Studer, Chem. Sci. 10 (2019) 8503–8518. doi: 10.1039/c9sc03765a
M. Yan, J.C. Lo, J.T. Edwards, P.S. Baran, J. Am. Chem. Soc. 138 (2016) 12692–12714. doi: 10.1021/jacs.6b08856
Y. Li, L. Ge, M.T. Muhammad, H. Bao, Synthesis49 (2017) 5263–5284. doi: 10.1055/s-0036-1590935
T. Patra, D. Maiti, Chem. Eur. J. 23 (2017) 7382–7401. doi: 10.1002/chem.201604496
S. Murarka, Adv. Synth. Catal. 360 (2018) 1735–1753. doi: 10.1002/adsc.201701615
P. Niu, J. Li, Y. Zhang, C. Huo, Eur. J. Org. Chem. 36 (2020) 5801–5814. doi: 10.1002/ejoc.202000525
W. M. Cheng, R. Shang, ACS Catal. 10 (2020) 9170–9196. doi: 10.1021/acscatal.0c01979
S.K. Parida, T. Mandal, S. Das, et al., ACS Catal. 11 (2021) 1640–1683. doi: 10.1021/acscatal.0c04756
L. Xu, Eur. J. Org. Chem. 29 (2018) 3884–3890. doi: 10.1002/ejoc.201800534
D. Wei, T.M. Liu, B. Zhou, B. Han, Org. Lett. 22 (2020) 234–238. doi: 10.1021/acs.orglett.9b04218
L. Candish, M. Teders, F. Glorius, J. Am. Chem. Soc. 139 (2017) 7440–7443. doi: 10.1021/jacs.7b03127
W.M. Cheng, R. Shang, B. Zhao, W.L. Xing, Y. Fu, Org. Lett. 19 (2017) 4291–4294. doi: 10.1021/acs.orglett.7b01950
C. Li, J. Wang, L.M. Barton, et al., Science356 (2017) eaam7355. doi: 10.1126/science.aam7355
J. Wang, M. Shang, H. Lundberg, et al., ACS Catal. 8 (2018) 9537–9542. doi: 10.1021/acscatal.8b02928
D. Hu, L. Wang, P. Li, Org. Lett. 19 (2017) 2770–2773. doi: 10.1021/acs.orglett.7b01181
A. Fawcett, J. Pradeilles, Y. Wang, et al., Science357 (2017) 283–286. doi: 10.1126/science.aan3679
J.I. Yo-shida, K. Kataoka, R. Horcajada, A. Nagaki, Chem. Rev. 108 (2008) 2265–2299. doi: 10.1021/cr0680843
B.A. Frontana-Uribe, R.D. Little, J.G. Ibanez, A. Palma, R. Vasquez-Medrano, Green Chem. 12 (2010) 2099–2119. doi: 10.1039/c0gc00382d
R. Francke, R.D. Little, Chem. Soc. Rev. 43 (2014) 2492–2521. doi: 10.1039/c3cs60464k
S.R. Waldvogel, B. Janza, Angew. Chem. Int. Ed. 53 (2014) 7122–7123. doi: 10.1002/anie.201405082
E.J. Horn, B. R. Rosen, P.S. Baran, ACS Cent. Sci. 2 (2016) 302–308. doi: 10.1021/acscentsci.6b00091
M. Yan, Y. Kawamata, P.S. Baran, Chem. Rev. 117 (2017) 13230–13319. doi: 10.1021/acs.chemrev.7b00397
A. Wiebe, T. Gieshoff, S. Möhle, et al., Angew. Chem. Int. Ed. 57 (2018) 5594–5619. doi: 10.1002/anie.201711060
D. Pollok, S.R. Waldvogel, Chem. Sci. 11 (2020) 12386–12400. doi: 10.1039/d0sc01848a
X. Chen, X. Luo, X. Peng, et al., Chem. Eur. J. 26 (2020) 3226–3230. doi: 10.1002/chem.201905224
Y. Liu, L. Xue, B. Shi, et al., Chem. Commun. 55 (2019) 14922–14925. doi: 10.1039/c9cc08528a
F. Lian, K. Xu, W. Meng, et al., Chem. Commun. 55 (2019) 14685–14688. doi: 10.1039/c9cc07840a
J. Hong, Q. Liu, F. Li, et al., Chin. J. Chem. 37 (2019) 347–351. doi: 10.1002/cjoc.201900001
L.M. Barton, L. Chen, D. Blackmond, P. Baran, Proc. Natl. Acad. Sci. U. S. A. 118 (2021) e2109408118. doi: 10.1073/pnas.2109408118

Scheme 2 Substrate scope. Reaction conditions: NHPI ester (0.6 mmol, 1 equiv.), B2cat2 (2 equiv.), DMF (2 mL), platinum plate anode: (10 mm × 10 mm × 0.2 mm), platinum plate cathode: (10 mm × 10 mm × 0.2 mm), nBu4NBF4 (0.6 mol/L), constant current: 10 mA, r.t., argon atmosphere, 4 h, undivided cell, then pinacol (4 equiv.), Et3N (2 mL), r.t., 2 h. aReaction time was 3 h. bReaction time was 6 h. cB2cat2 (3 equiv.) was used.

Scheme 3 Control experiments and proposed mechanism for the electrochemical decarboxylative borylation reaction.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:


 下载:
下载:
 下载:
下载: