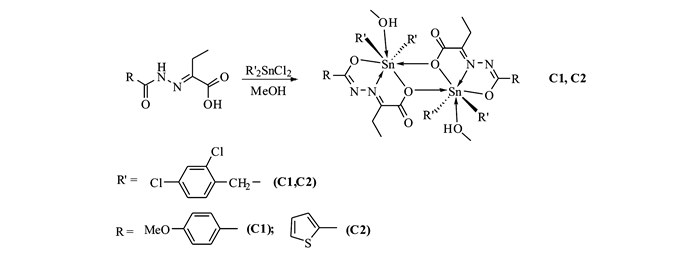
Scheme 1.
Synthetic route of complexes C1 and C2

癌症作为导致人类疾病死亡的原因之一,严重威胁着人类的生命健康。早期治疗癌症的化疗药物主要有顺铂、卡铂和奥沙利铂等铂类药物,但是,在后期的使用过程中,这类药物暴露出的高毒性、耐药性等问题[1-3]迫使人们开发并研究新型的抗癌药物[4-6]。自1980年Crowe等[7]首次报道二烃基锡化合物具有抑制癌细胞增殖作用以来,二烃基锡化合物在抗癌药物领域备受关注,目前,已有大量文献[8-10]报道二烃基锡化合物是最具有前景的金属抗癌药物之一。然而,常规二烃基锡化合物具有生物兼容性差,脂水分配系数大的缺点,严重制约着其药物的应用,因此,利用具有良好生物兼容性的配体同二烃基锡反应制备新型有机锡配合物并研究其抗癌活性方面的工作就尤为重要。基于本课题组前期的研究基础[11-14],本文选择具有生物兼容性及复杂多变配位模式的酰腙配体与二(2, 4-二氯苄基)二氯化锡反应,合成了两个二烃基锡配合物C1和C2(Scheme 1),初步研究配合物对癌细胞的抑制效果以及与小牛胸腺DNA的相互作用机制,为筛选具有高抗癌活性的新型有机金属配合物奠定基础。
对甲氧基苯甲酰肼缩2-丁酮酸、2-噻吩酰肼缩2-丁酮酸酸和二(2, 4-二氯苄基)二氯化锡参考文献[15-16]方法合成;溴化乙锭(EB)、小牛胸腺DNA、三羟甲基氨基甲烷(Tris)购自Sigma-Aldrich公司;卡铂购自百灵威科技有限公司;人肺癌细胞(NCI-H460)、人肝癌细胞(HepG2)和人乳腺癌细胞(MCF7)细胞株购自美国组织培养库(ATCC);含10%胎牛血清的RPMI 1640培养基购自美国GIBICO公司;其它试剂均为分析纯。Tris-HCl (0.01 mol/L)缓冲溶液通过称取一定量Tris用0.1 mol/L的盐酸溶液调至pH=7.40,使用前配制;小牛胸腺DNA的纯度通过比较260和280 nm处的吸光度来确定(A260:A280=1.8:1~1.9:1),用所需pH值条件下缓冲溶液配制,浓度通过测定260 nm处的吸光度计算而得(ε260=6600 L/(mol·cm)),其储备液置于4 ℃保存;溴化乙锭溶液用pH=7.40的Tris-HCl(0.01 mol/L)缓冲溶液配制。
Prestige-21型傅里叶变换红外光谱仪(FTIR,日本岛津公司);Bruker AVANCE-500型核磁共振波谱仪(NMR,德国布鲁克公司);Bruker SMART APEX Ⅱ CCD型单晶衍射仪(SCXRD,德国布鲁克公司);Thermo Scientific LTQ Orbitrap XL型高分辨质谱(HRMS,美国赛默飞世尔公司);NETZSCH TG 209 F3型热重分析仪(TGA,德国耐驰公司);F-7000型荧光光谱仪(日本日立公司);UV-2550型紫外-可见分光光度计(UV-Vis,日本岛津公司);MOS-500型圆二色光谱仪(CD,法国比奥罗杰公司);X-4型双目体视显微熔点测定仪(北京泰克仪器有限公司)。
于50 mL圆底烧瓶中,加入1 mmol对甲氧基苯甲酰肼缩2-丁酮酸或2-噻吩酰肼缩2-丁酮酸,1 mmol二(2, 4-二氯苄基)二氯化锡,25 mL无水甲醇,搅拌回流4 h。冷却,过滤,旋转蒸除溶剂,用甲醇重结晶,得淡黄色晶体C1或C2。配合物C1和C2的IR谱图、13C NMR图、119Sn图和HRM图见辅助材料图S1-S10。
配合物C1:淡黄色晶体,产率72%。mp 176~178 ℃,FTIR(KBr), σ/cm-1:3080, 2974, 2936, 2828, 1676, 1632, 1603, 1584, 1514, 1472, 1389, 1339, 1302, 1290, 1252, 1169, 1103, 1092, 1063, 1040, 1024, 970, 841, 816, 804, 762, 725, 710, 700, 627, 592, 532, 511, 449;1H NMR(500 MHz, CDCl3), δ:7.84(d, J=8.7 Hz, 2H), 7.21(s, 2H), 7.04(d, J=8.5 Hz, 2H), 6.98(dd, J1=8.2 Hz, J2=1.8 Hz, 2H), 6.89(d, J=8.7 Hz, 2H), 3.88(s, 3H), 3.22(d, J=12.2 Hz, 2H), 3.13(d, J=12.2 Hz, 2H), 2.94(q, J=7.3 Hz, 2H), 1.16(t, J=7.3 Hz, 3H); 13C NMR(126 MHz, CDCl3), δ:174.31, 163.95, 163.24, 157.91, 133.37, 133.23, 132.11, 131.01, 130.49, 128.82, 127.28, 124.53, 113.62, 55.45, 50.84, 28.52, 20.75, 10.25;119Sn NMR(Me4Sn, 187 MHz, CDCl3), δ:-264.84;HRMS(ESI) m/z计算值C26H23Cl4N2O4Sn+ [M-CH3OH+H]+ 686.9428, 实测值686.9432。
配合物C2:黄色晶体,产率69%。mp 195~197 ℃,FTIR(KBr), σ/cm-1:3107, 2974, 2936, 2874, 2824, 1628, 1611, 1584, 1530, 1472, 1431, 1387, 1325, 1312, 1263, 1194, 1144, 1113, 1090, 1063, 1034, 858, 845, 816, 802, 752, 739, 727, 712, 656, 592, 559, 530, 498, 444;1H NMR(500 MHz, CDCl3), δ:7.59(dd, J1=3.7 Hz, J2=1.2 Hz, 1H), 7.53(dd, J1=4.8 Hz, J2=1.2 Hz, 1H), 7.20(d, J=2.1 Hz, 2H), 7.05~7.09(m, 3H), 6.96(dd, J1=8.2 Hz, J2=2.1 Hz, 2H), 3.28(d, J=12.3 Hz, 2H), 3.18(d, J=12.3 Hz, 2H), 2.90(q, J=7.6 Hz, 2H), 1.15(t, J=7.3 Hz, 3H); 13C NMR(126 MHz, CDCl3), δ:170.64, 164.09, 157.90, 136.34, 133.35, 133.14, 132.15, 132.01, 131.61, 131.19, 128.78, 127.72, 127.20, 50.85, 29.54, 20.78, 10.24;119Sn NMR(Me4Sn, 187 MHz, CDCl3), δ:-284.04;HRMS(ESI) m/z计算值C23H19Cl4N2O3SSn+ [M-CH3OH+H]+ 662.8887, 实测值662.8857。
分别选取0.24 mm×0.22 mm×0.22 mm配合物C1和0.22 mm×0.20 mm×0.20 mm配合物C2的晶体,在Bruker SMART APEX Ⅱ CCD单晶衍射仪上,采用经石墨单色化的MoKα射线(λ=0.071073 nm),以φ~ω扫描方式收集衍射数据。可观察衍射点[I>2σ(I)]用于结构分析和精修。全部数据经Lp因子和经验吸收校正。晶体结构由直接法解出,全部非氢原子坐标在差值Fourier合成中陆续确定,理论加氢法及差值Fourier合成给出氢原子在晶胞中的位置坐标。对氢原子和非氢原子分别采用各向同性和各向异性热参数进行全矩阵最小二乘法修正,全部结构分析计算工作采用SHELX-97程序系统完成[17]。晶体学数据见表 1。CCDC:C1, 1909490;C2, 1909491。
 下载:
导出CSV
下载:
导出CSV
| Complex | C1 | C2 |
| Empirical formula | C54H52Cl8N4O10Sn2 | C48H44Cl8N4O8S2Sn2 |
| Formula weight | 1 437.98 | 1 389.97 |
| T/K | 296(2) | 293(2) |
| λ/nm | 0.071 073 | 0.071 073 |
| Crystal system | Triclinic | Monoclinic |
| Space group | P-1 | P21/n |
| a/nm | 1.072 26(8) | 1.250 86(16) |
| b/nm | 1.133 24(8) | 1.729 1(2) |
| c/nm | 1.288 15(10) | 1.322 48(17) |
| α/(°) | 77.829 0(10) | 90 |
| β/(°) | 74.681 0(10) | 98.257(2) |
| γ/(°) | 78.269 0(10) | 90 |
| V/nm3 | 1.457 52(19) | 2.830 7(6) |
| Z | 1 | 2 |
| Dc/(Mg·m-3) | 1.638 | 1.631 |
| Absorption coefficient/mm-1 | 1.283 | 1.387 |
| F(000) | 720 | 1 384 |
| Crystal size/mm | 0.24×0.22×0.22 | 0.22×0.20×0.20 |
| θ range/(°) | 2.51~25.10 | 1.95~25.10 |
| Limiting indices | -12≤h≤12, -13≤k≤13, -15≤l≤15 | -14≤h≤14, -20≤k≤13, -15≤l≤15 |
| Reflections collected/unique | 14 946/5 196(Rint=0.016 2) | 14 160/5 024(Rint=0.021 0) |
| Completeness | 0.997 | 0.999 |
| Max. and min. transmission | 0.765 5 and 0.748 2 | 0.768 9 and 0.750 1 |
| Data/restraints/parameters | 5 196/7/359 | 5 024/14/398 |
| Goodness-of-fit on F2 | 1.078 | 1.055 |
| Final R indices [I>2σ(I)] | R1=0.032 6, wR2=0.085 5 | R1=0.037 4, wR2=0.100 0 |
| R indices(all data) | R1=0.034 3, wR2=0.086 7 | R1=0.049 2, wR2=0.106 5 |
| (Δρ)max/(e·nm-3) | 1 791 | 1 194 |
| (Δρ)min/(e·nm-3) | -747 | -735 |
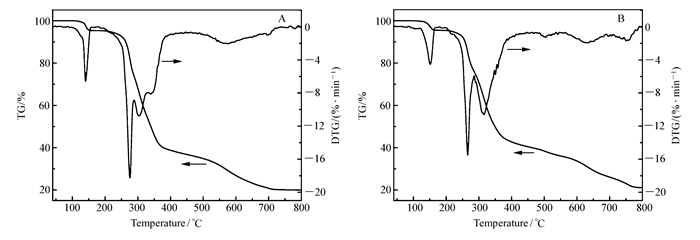
在空气氛下,加热速度为20 ℃/min,气体流速为20 mL/min的条件下,对配合物在40~800 ℃范围内进行了热重测试。
将待测药物溶于少量DMSO,用水稀释至所需浓度,保持最终DMSO浓度<0.1%。NCI-H460、HepG2和MCF7细胞用含10%胎牛血清的RPMI 1640(GIBICO公司)培养基,在5%(体积分数)CO2、37 ℃饱和湿度培养箱内进行体外培养。体外抗癌药敏试验是通过3-(4,5-二甲基噻唑-2)-2,5-二苯基四氮唑溴盐(MTT)法测定。数据处理使用Graph Pad Prism version 7.0程序,化合物半抑制浓度(IC50)通过程序中具有S形剂量响应的非线性回归模型进行拟合得到。
在5 mL容量瓶中分别加入配合物溶液(50 μmol/L)及不同浓度的ct-DNA(0~80 μmol/L),用Tris-HCl缓冲溶液定容,混匀,25 ℃下放置3.0 h,不同浓度的ct-DNA溶液为参比,分别扫描230~600 nm范围内的紫外-可见吸收光谱。
在5 mL容量瓶中分别加入ct-DNA(30 μmol/L)、EB(3 μmol/L)及不同浓度的配合物溶液(0~80 μmol/L),用Tris-HCl缓冲溶液定容,混匀,25 ℃下放置3.0 h,分别扫描荧光光谱,激发波长为258 nm,激发和发射光谱扫描狭缝宽度均为5.0 nm。
使用乌氏粘度计进行粘度测量,整个测量实验在温度恒定为(25.00±0.02) ℃的超级恒温水浴槽中进行。向乌氏粘度计中加入ct-DNA(50 μmol/L)及不同浓度的配合物溶液(0~50 μmol/L),用Tris-HCl缓冲溶液定容,混合均匀后,用精确度为0.01 s的电子秒表分别测定该溶液流经毛细管所需的时间,每次加液后平均测量时间3次,取平均值。按照式(1)计算相对粘度,
|
$ \eta = \left( {t - {t_0}} \right)/{t_0} $ |
(1) |
式中,t0为Tris-HCl缓冲溶液流经毛细管所需时间(s),t为ct-DNA和配合物的混合溶液流经毛细管所需时间(s),η0为没有加入配合物时ct-DNA溶液的相对粘度。以(η/η0)1/3对(ccomplex/cDNA)作图,得到不同浓度配合物对ct-DNA粘度的影响。
在5 mL容量瓶中分别加入配合物溶液(0或10 μmol/L)及ct-DNA(100 μmol/L),用Tris-HCl缓冲溶液定容,混匀,25 ℃下放置3.0 h。在220~350 nm范围内分别测定CD光谱。
在配合物C1和C2的红外谱图中,两个配合物分别在1584 cm-1处的吸收峰归属为酰腙(C=N—N=C)键的特征吸收[15, 18],分子中羧基的反对称伸缩振动峰和对称伸缩振动峰分别在1602、1388和1610、1386 cm-1处,二者频率之差分别为214、224 cm-1,表明两个配合物中的羧酸根均是以单齿形式与Sn发生配位[19]。此外,配合物C1和C2配位键的特征峰ν(Sn—O—Sn)、ν(Sn—O)、ν(Sn—N)和ν(Sn—C)分别位于592、532、511、449 cm-1和592、530、498、444 cm-1处,与文献[20-21]报道的类似化合物的出峰位置一致,由此初步表明有机锡配合物的形成。
在1H NMR谱中,其各组峰的积分面积之比与预期结构的各组质子数相对吻合[22]。从谱图中可以看到,配合物C1和C2中芳环上氢质子的出峰位置分别在6.89~7.85和6.95~7.60;2, 4-二氯苄基上亚甲基氢质子分别在3.13~3.22和3.18~3.28处出峰,呈现一组双重峰,推测可能是由于与锡相连的2, 4-二氯苄基空间位阻较大,导致其不能自由取向,使得亚甲基的两个质子发生同碳耦合,最终呈现双重峰;对甲氧基苯甲酰肼缩2-丁酮酸和2-噻吩酰肼缩2-丁酮酸配体部分的乙基氢质子分别在2.94、1.16和2.90、1.15处呈现四重峰和三重峰,两个配合物的所有氢质子出峰基本保持一致,说明了两个配合物具有相似的不对称结构单元。在13C NMR谱中,其各组峰与理论推测结构碳原子数相吻合[22]。配合物C1和C2中2, 4-二氯苄基上亚甲基碳原子的分别在28.52、29.54处呈现一个正常的单峰和一对小卫星峰,这是由于Sn—C耦合导致的。在119Sn NMR谱中,C1和C2分别在-264.84和-284.04处呈现一个单峰,表明两个配合物中均仅存在一种单一的有机锡化合物。
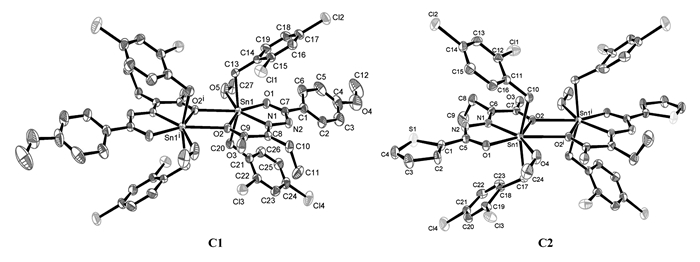
配合物C1、C2的主要键长和键角数据分别列于表 2,分子结构见图 2。配合物C1、C2均为双锡核分子,分子中心存在一个Sn2O2平面中心四元环,环的中心就是分子的对称中心,四元环由羧基氧原子以μ3-桥联配位Sn原子,且与两个锡原子的键长不等,其中Sn1—O2:0.2306(2) nm(C1);0.2307(3) nm(C2),属于正常Sn—O共价键长;而Sn1—O2i:0.26456(19) nm(C1);0.27104(27) nm(C2),虽大于锡氧两原子的共价半径之和,但小于锡原子与氧原子范氏半径之和,说明两个配合物中的Sn1与O2i之间均存在较强的作用[23]。
下载:
导出CSV
| C1 | |||||
| Sn(1)—C(13) | 0.214 4(3) | Sn(1)—C(20) | 0.215 1(3) | Sn(1)—O(1) | 0.215 9(2) |
| Sn(1)—N(1) | 0.221 9(2) | Sn(1)—O(2) | 0.230 6(2) | Sn(1)—O(5) | 0.254 8(3) |
| Sn(1)—O(2)i | 0.264 56(19) | ||||
| C(13)—Sn(1)—C(20) | 158.39(13) | C(13)—Sn(1)—O(1) | 93.92(11) | C(20)—Sn(1)—O(1) | 94.26(11) |
| C(13)—Sn(1)—N(1) | 102.62(11) | C(20)—Sn(1)—N(1) | 98.95(11) | O(1)—Sn(1)—N(1) | 70.90(8) |
| C(13)—Sn(1)—O(2) | 92.01(11) | C(20)—Sn(1)—O(2) | 94.03(11) | O(1)—Sn(1)—O(2) | 141.30(8) |
| N(1)—Sn(1)—O(2) | 70.49(8) | C(13)—Sn(1)—O(5) | 80.33(14) | C(20)—Sn(1)—O(5) | 82.79(14) |
| O(1)—Sn(1)—O(5) | 73.92(9) | N(1)—Sn(1)—O(5) | 144.81(10) | O(2)—Sn(1)—O(5) | 144.69(9) |
| C(13)—Sn(1)—O(2)i | 80.850(107) | C(20)—Sn(1)—O(2)i | 82.810(102) | O(2)—Sn(1)—O(2)i | 65.606(71) |
| O(5)—Sn(1)—O(2)i | 79.134(92) | ||||
| C2 | |||||
| Sn(1)—C(17) | 0.213 7(4) | Sn(1)—C(10) | 0.214 0(4) | Sn(1)—O(1) | 0.215 3(3) |
| Sn(1)—N(1) | 0.222 4(3) | Sn(1)—O(2) | 0.230 7(3) | Sn(1)—O(4) | 0.253 3(4) |
| Sn(1)—O(2)i | 0.271 04(27) | ||||
| C(17)—Sn(1)—C(10) | 158.05(17) | C(17)—Sn(1)—O(1) | 94.22(15) | C(10)—Sn(1)—O(1) | 96.04(14) |
| C(17)—Sn(1)—N(1) | 102.18(15) | C(10)—Sn(1)—N(1) | 99.50(15) | O(1)—Sn(1)—N(1) | 71.08(11) |
| C(17)—Sn(1)—O(2) | 92.15(14) | C(10)—Sn(1)—O(2) | 91.82(14) | O(1)—Sn(1)—O(2) | 141.52(10) |
| N(1)—Sn(1)—O(2) | 70.47(11) | C(17)—Sn(1)—O(4) | 83.65(17) | C(10)—Sn(1)—O(4) | 80.19(17) |
| O(1)—Sn(1)—O(4) | 75.61(12) | N(1)—Sn(1)—O(4) | 146.49(13) | O(2)—Sn(1)—O(4) | 142.86(12) |
| C(10)—Sn(1)—O(2)i | 81.279(142) | C(17)—Sn(1)—O(2)i | 80.940(132) | O(2)—Sn(1)—O(2)i | 64.714(87) |
| O(4)—Sn(1)—O(2)i | 78.222(102) | ||||
| Symmetry code for complex C1:i 2-x, 2-y, 1-z; C2:i1-x, 2-y, -z. | |||||
Symmetry code for C1: i2-x, 2-y, 1-z; C2:i1-x, 2-y, -z
配合物C1的中心锡原子Sn1与来自配体中的两个氧原子O1和O2,一个亚氨基氮原子N1,一个配位甲醇氧原子O5,来自两个对甲基苄基中的亚甲基碳原子C13和C20以及来自另一个配体分子中的O2i等配位,形成七配位五角双锥构型。O1、O2、O5、N1、O2i占据了赤道平面的5个位置,两个亚甲基碳原子C13和C20则占据了该平面两侧的轴向位置,轴向C13—Sn1—C20键角为158.39(13)°,与180°偏离了21.61°,且赤道平面的5个原子与中心锡原子的键长不等(dSn1—O1=0.2159(2) nm;dSn1—O2=0.2306(2) nm;dSn1—O5=0.2548(3) nm;dSn1—N1=0.2219(2) nm;dSn1—O2i=0.26456(19) nm),且键角也不相等(∠O1—Sn1—O5 73.92(9)°;∠O1—Sn1—N1 70.90(8)°;∠N1—Sn1—O2 70.49(8)°;∠O2—Sn1—O2i 65.606(71)°;∠O5—Sn1—O2i 79.134(92)°),因此,该配合物中心锡原子为七配位畸变五角双锥构型。
配合物C2与配合物C1相似,中心锡原子Sn1也是形成七配位五角双锥构型。轴向C10—Sn1—C17键角为158.05(17)°,与180°偏离了22.95°,比配合物C1的偏离程度大,在配合物C2中,赤道平面的5个原子与中心锡原子的键长也均不相等,因此,配合物C2中心锡原子也为七配位畸变五角双锥构型。
配合物C1和C2的热稳定曲线如图 3所示,随温度的升高,配合物发生相似的失重过程。在初始阶段40~200 ℃,配合物C1失重为4.63%(理论值:4.45%),C2为4.46%(理论值:4.60%),分别对应配合物失去配位的甲醇分子;配合物C1、C2的中间失重阶段均相对模糊,在200~800 ℃范围内失重,对应配合物分子分别失去对甲氧基苯甲酰肼缩2-丁酮酸或2-噻吩酰肼缩2-丁酮酸配体及2, 4-二氯苄基,最终稳定在约19.98%(C1)和21.11%(C2),残余物与SnO2的计算含量20.86%(C1)及21.58%(C2)吻合;上述结果表明配合物C1、C2均在120 ℃之前可稳定存在。
表 3分别列出了配合物C1、C2和卡铂对体外培养癌细胞NCI-H460、HepG2、MCF7的抑制活性。配合物C1、C2对3种癌细胞均有一定的抑制作用。配合物C1对3种癌细胞的抑制活性均优于卡铂,对HepG2癌细胞最为敏感,其IC50=(3.00±0.11) μmol/L。配合物C2对HepG2和MCF7癌细胞的抑制活性也强于卡铂,但是对NCI-H460细胞的抑制效果相对卡铂较差。总体而言,配合物C1的抗癌活性略优于C2。
下载:
导出CSV
| IC50/(μmol·L-1) | |||
| NCI-H460 | HepG2 | MCF7 | |
| C1 | 5.34±0.37 | 3.00±0.11 | 4.57±0.12 |
| C2 | 8.41±0.25 | 7.27±0.34 | 5.20±0.18 |
| Carboplatin | 7.26±0.32 | 7.70±0.25 | 8.22±0.41 |
FTIR、NMR、X-ray等的表征数据表明,配合物C1、C2结构非常相似,中心锡原子的配位模式和配位数也是相同的,仅在酰腙配体部分的芳环有所不同,配合物C1中的芳环为对甲氧基苯基,配合物C2中的为噻吩环,由此说明配体芳环的不同对配合物的抗癌活性虽有影响,但是效果不明显,因而推测,酰腙配体部分的芳环取代基不是关键的药效团,有机锡配合物表现出的抗癌活性与配体和烃机锡的协同作用有关[24-25]。
为了准确表达配合物与DNA相互作用的强度,可以根据式(2)计算的二者间的结合常数(Kb)[26]:
|
$ {c_{{\rm{DNA}}}}/\left( {{\varepsilon _{\rm{A}}} - {\varepsilon _{\rm{F}}}} \right) = {c_{{\rm{DNA}}}}/\left( {{\varepsilon _{\rm{B}}} - {\varepsilon _{\rm{F}}}} \right) + 1/{K_{\rm{b}}}\left( {{\varepsilon _{\rm{B}}} - {\varepsilon _{\rm{F}}}} \right) $ |
(2) |
式中,εA、εF、εB分别为任意DNA浓度下溶液的摩尔消光系数、自由配合物的摩尔消光系数、配合物被DNA完全键合时的摩尔消光系数。根据方程式线性拟合,通过斜率和截距计算出配合物C1和C2的Kb分别为5.8×103 L/mol(C1)和4.3×103 L/mol (C2),两数值略有差异,但还是足以说明配合物C1与DNA的作用略强于配合物C2。并且,该Kb与文献[27-28]报道的类似配合物与DNA作用的结合常数大小相近,表明配合物C1和C2均与DNA的作用较强。
从图 4中可以看出,当配合物C1和C2与DNA发生作用时,它们的紫外-可见光谱吸收峰均表现出了减色和红移现象,配合物C1减色率为29.1%,红移5 nm,配合物C2减色率为22.0%,红移5 nm,减色越明显表明配合物与DNA相互作用越强[26]。出现这种现象的原因可能是配合物通过嵌入作用与DNA结合后与碱基对发生π电子堆积,配体的π*空轨道与DNA碱基对的π轨道发生偶合导致能级下降,偶合后的π*轨道部分填充电子,使其π-π*跃迁几率减小,从而产生减色效应。上述结果表明,配合物C1和C2均可通过嵌入作用与双链ct-DNA结合。
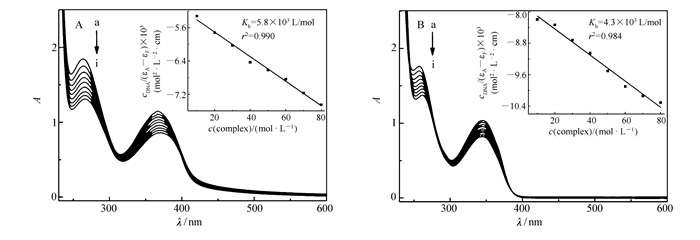
ccomplex=50 μmol/L; from a to i, cDNA=0, 10, 20, 30, 40, 50, 60, 70, 80 μmol/L, respectively
Inset:plots of cDNA/(εA-εF) vs ccomplex
图 5显示,在596 nm波长下,随着配合物C1或C2浓度的增加,配合物和EB-DNA体系的荧光强度明显减弱。由此说明,配合物C1或C2和EB竞争与DNA结合,使体系中的EB游离出来,从而导致体系荧光强度减弱,其作用方式可能为嵌插作用。而且根据经典Stern-Volmer方程[25]:I0/I=1+KSVccomplex,由曲线拟合计算出配合物C1和C2与DNA作用的猝灭常数(KSV)分别为3.7×104和2.8×104 L/mol,比文献[29-30]报道的结合常数略大,表明配合物与DNA存在较强的插入作用,单独比较配合物C1和C2的猝灭常数,结果表明配合物C1与DNA作用略强于配合物C2,与紫外-可见光谱所得结论保持一致。
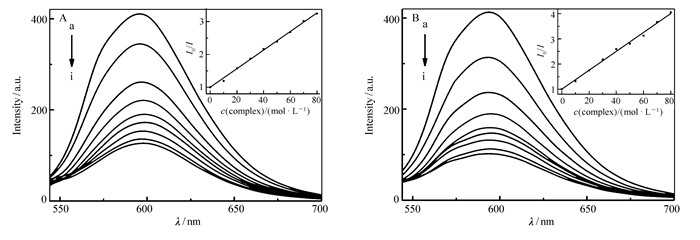
cDNA=30 μmol/L; cEB=3 μmol/L; from a to i, ccomplex=0, 10, 20, 30, 40, 50, 60, 70, 80 μmol/L, respectively. Inset:plots of I0/I vs ccomplex, λex=258 nm
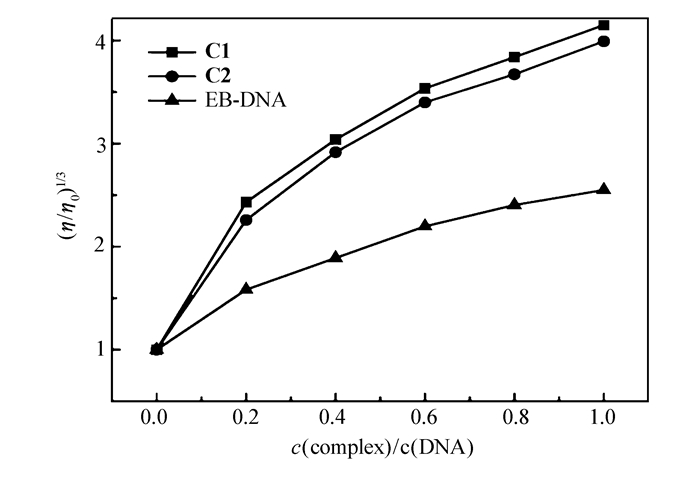
粘度测试是研究配合物与DNA作用方式的方法之一。配合物与ct-DNA以嵌入方式相互作用时,DNA双螺旋链增长,DNA粘度增大;以部分插入的方式作用时,DNA溶液的粘度降低;以静电或沟面方式作用时,DNA溶液的粘度未发生明显变化[31]。溴化乙锭(EB)是典型的DNA嵌入剂,与DNA发生作用时会使其粘度增加。图 6为DNA在不同浓度配合物C1、C2和EB溶液存在下粘度的变化趋势,可以看出,加入配合物C1、C2后,DNA的相对粘度总体上均是随着配合物浓度的增大呈现上升的趋势,类似于EB,说明配合物以嵌入的方式与DNA结合,该结论与其它光谱数据所得结论一致。
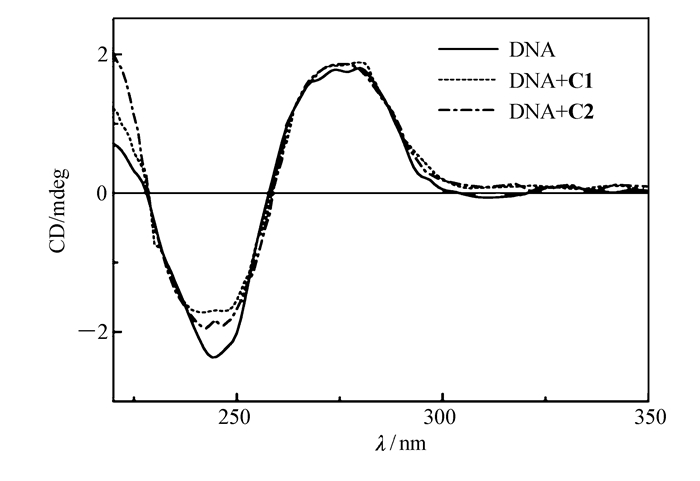
圆二色光谱常用来研究配合物与DNA的相互作用,一般B型DNA较为常见,其正吸收峰出现在275 nm附近,与DNA的碱基堆积有关;负峰出现在245 nm附近,与DNA的右手螺旋性质有关[32-33]。图 7为配合物C1、C2与ct-DNA相互作用的圆二色光谱图。配合物C1、C2的变化趋势相似,加入配合物之后,ct-DNA的负峰强度(245 nm附近的峰强度)明显下降,说明加入配合物之后影响了DNA的右手螺旋性质;而加入配合物之后,DNA在275 nm处的正峰信号变化并不明显,说明加入配合物对ct-DNA的碱基堆积作用也存在一定的影响,但是影响不大。上述结果表明,配合物C1、C2均可以与DNA发生相互作用,对DNA的右手螺旋性质产生影响。
二(2, 4-二氯苄基)二氯化锡分别与对甲氧基苯甲酰肼缩2-丁酮酸或2-噻吩酰肼缩2-丁酮酸反应,合成了两个二(2, 4-二氯苄基)锡配合物(C1、C2)。配合物C1和C2均为双锡核分子,以Sn2O2四元环为中心对称,且锡原子与配位原子形成七配位畸变五角双锥构型。在空气氛下,配合物C1和C2均在120 ℃以下可稳定存在。配合物C1和C2对人肺癌细胞(NCI-H460)、人肝癌细胞(HepG2)和人乳腺癌细胞(MCF7)都有较好的抑制作用,但是总体而言,配合物C1略优于配合物C2。配合物C1、C2与小牛胸腺DNA的相互作用方式是插入结合,且配合物C1与DNA的作用强于配合物C2。
辅助材料(Supporting Information)[配合物C1和C2的FTIR、NMR和HRMS]可以免费从本刊网站(http://yyhx.ciac.jl.cn/)下载。
Shahid F, Farooqui Z, Khan F. Cisplatin-induced Gastrointestinal Toxicity:An Update on Possible Mechanisms and on Available Gastroprotective Strategies[J]. Eur J Pharmacol, 2018, 827: 49-57. doi: 10.1016/j.ejphar.2018.03.009
Karasawa T, Steyger P S. An Integrated View of Cisplatin-induced Nephrotoxicity and Ototoxicity[J]. Toxicol Lett, 2015, 237(3): 219-227. doi: 10.1016/j.toxlet.2015.06.012
Milosavljevic N, Duranton C, Djerbi N. Nongenomic Effects of Cisplatin:Acute Inhibition of Mechanosensitive Transporters and Channels Without Actin Remodeling[J]. Cancer Res, 2010, 70(19): 7514-7522. doi: 10.1158/0008-5472.CAN-10-1253
Cao W, Qi J, Qian K. Structure-activity Relationships of 2-Quinolinecarboxaldehyde Thiosemicarbazone Gallium(Ⅲ) Complexes with Potent and Selective Anticancer Activity[J]. J Inorg Biochem, 2019, 191: 174-182. doi: 10.1016/j.jinorgbio.2018.11.017
Zaki M, Hairat S, Aazam E S. Scope of Organometallic Compounds Based on Transition Metal-Arene Systems as Anticancer Agents:Starting from the Classical Paradigm to Targeting Multiple Strategies[J]. RSC Adv, 2019, 9(6): 3239-3278. doi: 10.1039/C8RA07926A
Kenny R G, Marmion C J. Toward Multi-targeted Platinum and Ruthenium Drugs-A New Paradigm in Cancer Drug Treatment Regimens[J]. Chem Rev, 2019, 119(2): 1058-1137.
Crowe A J, Smith P J, Atassi G. Investigations into the Antitumour Activity of Organotin Compounds. I.Diorganotin Dihalide and Di-pseudohalide Complexes[J]. Chem-Biol Interact, 1980, 32(1): 171-178.
Attanzio A, Ippolito M, Girasolo M A. Anti-cancer Activity of Di- and Tri-organotin(Ⅳ) Compounds with D-(+)-Galacturonic Acid on Human Tumor Cells[J]. J Inorg Biochem, 2018, 188: 102-112. doi: 10.1016/j.jinorgbio.2018.04.006
Shang X, Meng X, Alegria E C B A. Syntheses, Molecular Structures, Electrochemical Behavior, Theoretical Study, and Antitumor Activities of Organotin(Ⅳ) Complexes Containing 1-(4-Chlorophenyl)-1-cyclopentanecarboxylato Ligands[J]. Inorg Chem, 2011, 50(17): 8158-8167. doi: 10.1021/ic200635g
Basu Baul T S, Paul A, Pellerito L. Dibutyltin(Ⅳ) Complexes Containing Arylazobenzoate Ligands:Chemistry, in Vitro Cytotoxic Effects on Human Tumor Cell Lines and Mode of Interaction with Some Enzymes[J]. Invest New Drug, 2011, 29(2): 285-299. doi: 10.1007/s10637-009-9360-3
Tan Y X, Zhang Z J, Feng Y L. Syntheses, Crystal Structures and Biological Activity of the 1D Chain Benzyltin Complexes Based on 2-Oxo-Propionic Acid Benzoyl Hydrazone[J]. J Inorg Organomet P, 2017, 27(1): 342-352. doi: 10.1007/s10904-016-0477-5
Tan Y, Zhang Z, Tan Y. Syntheses, Crystal Structures and Biological Activity of the Dialkytin Complexes Based on 2-Oxo-3-phenylpropionic Acid Salicyloylhydrazone[J]. J Coord Chem, 2017, 70(15): 2606-2624. doi: 10.1080/00958972.2017.1355460
郑建华, 刘俊, 肖尧. 基于水杨酰腙配体的二丁基锡配合物的合成、晶体结构、热稳定性及与DNA相互作用[J]. 应用化学, 2015,32,(5): 562-569. ZHENG Jianhua, LIU Jun, XIAO Yao. Synthesis, Crystal Structure, Thermal Stability and DNA Interaction of the Dibutytin Complex Based on Salicylacylhydrazone[J]. Chinese J Appl Chem, 2015, 32(5): 562-569.
张志坚, 蒋伍玖, 刘洋. N-(2-丙酸)-芳甲酰腙二对甲基苄基锡配合物的合成、晶体结构及生物活性[J]. 无机化学学报, 2017,33,(9): 1603-1610. ZHANG Zhijian, JIANG Wujiu, LIU Yang. Syntheses, Crystal Structures and Biological Activity of N-(2-Propionic acid)-aroyl Hydrazone Di-p-methylbenzytin Complexes[J]. Chinese J Inorg Chem, 2017, 33(9): 1603-1610.
何水样, 曹文凯, 陈军利. 铜(Ⅱ)与2-羰基丙酸水杨酰腙配合物的合成、晶体结构和抑菌活性[J]. 高等学校化学学报, 2002,23,(6): 991-995. doi: 10.3321/j.issn:0251-0790.2002.06.002HE Shuiyang, CAO Wenkai, CHEN Junli. Synthesis, Crystal Structure and Antibacterial Activity of Copper(Ⅱ) Complexes with 2-Oxo-Propionic Acid Salicyloyl Hydrazone[J]. Chem J Chinese Univ, 2002, 23(6): 991-995. doi: 10.3321/j.issn:0251-0790.2002.06.002
Sisido K, Takeda Y, Kinugawa Z. Direct Synthesis of Organotin Compounds. I.Di- and Tribenzyltin Chlorides[J]. J Am Chem Soc, 1961, 83(3): 538-541. doi: 10.1021/ja01464a008
Sheldrick G M. SHELXL-97, A Program for Crystal Structure Refinement[M]. Germany:University of Ge ttingen, 1997.
García-López M C, Muñoz-Flores B M, Chan-Navarro R. Microwave-Assisted Synthesis, Third-Order Nonlinear Pptical Properties, Voltammetry Cyclic and Theoretical Calculations of Organotin Compounds Bearing Push-Pull Schiff Bases[J]. J Organomet Chem, 2016, 806: 68-76. doi: 10.1016/j.jorganchem.2016.01.030
Tian L J, Yao Y Z, Liu Q T. Synthesis, Characterization and Antibacterial Activity of Cyclohexyltin Complexes of N-(3, 5-Dibromosalicylidene)valine[J]. Chinese J Struct Chem, 2016, 35(6): 849-856.
Yang Y, Hong M, Xu L. Organotin(Ⅳ) Complexes Derived from Schiff Base N'-[(1E)-(2-hydroxy-3-methoxyphenyl)methylidene]pyridine-3-carbohydrazone:Synthesis, in Vitro Cytotoxicities and DNA/BSA Interaction[J]. J Organomet Chem, 2016, 804: 48-58. doi: 10.1016/j.jorganchem.2015.12.041
Barba V, Zaragoza J, Höpfl H. Use of Bis-aminoalcohol Benzoquinones and Dihydroxybenzoquinones in the Formation of Mono and Polymeric Structures of Diorganotin(Ⅳ) Derivatives[J]. J Organomet Chem, 2011, 696: 1949-1956. doi: 10.1016/j.jorganchem.2010.10.040
Pretsch E, Bühlmann P, Badertscher M. Structure Determination of Organic Compounds[M]. Fourth ed, Berlin Heidelberg:Springer-Verlag, 2009.
Wagner M, Zobel B, Dietz C. Cyclic Dinuclear Organotin Cations Stabilized by Bulky Substituents[J]. Organometallics, 2015, 34: 5602-5608. doi: 10.1021/acs.organomet.5b00829
Tan Y X, Zhang Z J, Feng Y L. Syntheses, Crystal Structures and Biological Activities of the 2-Oxo-butyric Acid Salicylacylhydrazone Dibenzyltin Complexes[J]. Chinese J Struct Chem, 2017, 36(6): 925-936.
Yan C, Zhang J, Liang T. Diorganotin(Ⅳ) Complexes with 4-Nitro-N-phthaloyl-glycine:Synthesis, Characterization, Antitumor Activity and DNA-Binding Studies[J]. Biomed Pharmacother, 2015, 71: 119-127. doi: 10.1016/j.biopha.2015.02.027
Pyle A M, Rehmann J P, Meshoyrer R. Mixed-ligand Complexes of Ruthenium(Ⅱ):Factors Governing Binding to DNA[J]. J Am Chem Soc, 1989, 111(8): 3051-3058. doi: 10.1021/ja00190a046
Liu K, Yan H, Chang G. Organotin(Ⅳ) Complexes Derived from Hydrazone Schiff Base:Synthesis, Crystal Structure, in Vitro Cytotoxicity and DNA/BSA Interactions[J]. Inorg Chim Acta, 2017, 464: 137-146. doi: 10.1016/j.ica.2017.05.017
Li S T, Ma Z Y, Liu X. Synthesis, Crystal Structures, DNA/bovine Serum Albumin Binding, DNA Cleavage and Cytotoxicity of Five Mononuclear Zinc(Ⅱ) Complexes[J]. Appl Organomet Chem, 2017, 31(11): e3802. doi: 10.1002/aoc.3802
Ganji N, Rambabu A, Vamsikrishna N. Copper(Ⅱ) Complexes with Isoxazole Schiff Bases:Synthesis, Spectroscopic Investigation, DNA Binding and Nuclease Activities, Antioxidant and Antimicrobial Studies[J]. J Mol Struct, 2018, 1173: 173-182. doi: 10.1016/j.molstruc.2018.06.100
Rahban M, Divsalar A, Saboury A A. Nanotoxicity and Spectroscopy Studies of Silver Nanoparticle:Calf Thymus DNA and K562 as Targets[J]. J Phys Chem C, 2010, 114(13): 5798-5803. doi: 10.1021/jp910656g
Zhao Y, Li Z, Li H. Synthesis, Crystal Structure, DNA Binding and in Vitro Cytotoxicity Studies of Zn(Ⅱ) Complexes Derived From Amino-Alcohol Schiff-Bases[J]. Inorg Chim Acta, 2018, 482: 136-143. doi: 10.1016/j.ica.2018.06.008
Chen Z F, Wei J H, Liu Y C. High Antitumor Activity of 5, 7-Dihalo-8-quinolinolato Cerium Complexes[J]. Eur J Med Chem, 2013, 68: 454-462. doi: 10.1016/j.ejmech.2013.08.007
Arjmand F, Sayeed F, Parveen S. In Vitro Binding Studies of Organotin(Ⅳ) Complexes of 1, 2-Bis(1H-benzimidazol-2-yl)ethane-1, 2-diol with CT-DNA and Nucleotides (5'-GMP and 5'-TMP):Effect of the Ancillary Ligand on the Binding Propensity[J]. J Organomet Chem, 2011, 696(24): 3836-3845. doi: 10.1016/j.jorganchem.2011.08.007

图 2 配合物C1、C2的分子结构图
Figure 2 Molecular structures of complexes C1 and C2
Symmetry code for C1: i2-x, 2-y, 1-z; C2:i1-x, 2-y, -z

图 4 配合物C1(A)和C2(B)与DNA相互作用的紫外可见光谱图
Figure 4 UV-Vis spectra of C1(A) and C2(B) upon addition of ct-DNA
ccomplex=50 μmol/L; from a to i, cDNA=0, 10, 20, 30, 40, 50, 60, 70, 80 μmol/L, respectively
Inset:plots of cDNA/(εA-εF) vs ccomplex

图 5 配合物C1(A)和C2(B)与EB-DNA体系相互作用的荧光光谱图
Figure 5 Effects of complex C1(A) and C2(B) on the fluorescent spectra of EB-DNA system
cDNA=30 μmol/L; cEB=3 μmol/L; from a to i, ccomplex=0, 10, 20, 30, 40, 50, 60, 70, 80 μmol/L, respectively. Inset:plots of I0/I vs ccomplex, λex=258 nm

图 6 不同浓度配合物C1、C2和EB对ct-DNA粘度的影响
Figure 6 Effect of increasing amounts of the complex C1, C2 and EB on the relative viscosity

图 7 配合物C1、C2与ct-DNA相互作用的圆二色光谱图
Figure 7 Circular dichroism spectra of ct-DNA in the absence and presence of complexes C1 or C2
表 1 配合物的晶体学参数
Table 1. Crystal parameters of synthesized complexes
| Complex | C1 | C2 |
| Empirical formula | C54H52Cl8N4O10Sn2 | C48H44Cl8N4O8S2Sn2 |
| Formula weight | 1 437.98 | 1 389.97 |
| T/K | 296(2) | 293(2) |
| λ/nm | 0.071 073 | 0.071 073 |
| Crystal system | Triclinic | Monoclinic |
| Space group | P-1 | P21/n |
| a/nm | 1.072 26(8) | 1.250 86(16) |
| b/nm | 1.133 24(8) | 1.729 1(2) |
| c/nm | 1.288 15(10) | 1.322 48(17) |
| α/(°) | 77.829 0(10) | 90 |
| β/(°) | 74.681 0(10) | 98.257(2) |
| γ/(°) | 78.269 0(10) | 90 |
| V/nm3 | 1.457 52(19) | 2.830 7(6) |
| Z | 1 | 2 |
| Dc/(Mg·m-3) | 1.638 | 1.631 |
| Absorption coefficient/mm-1 | 1.283 | 1.387 |
| F(000) | 720 | 1 384 |
| Crystal size/mm | 0.24×0.22×0.22 | 0.22×0.20×0.20 |
| θ range/(°) | 2.51~25.10 | 1.95~25.10 |
| Limiting indices | -12≤h≤12, -13≤k≤13, -15≤l≤15 | -14≤h≤14, -20≤k≤13, -15≤l≤15 |
| Reflections collected/unique | 14 946/5 196(Rint=0.016 2) | 14 160/5 024(Rint=0.021 0) |
| Completeness | 0.997 | 0.999 |
| Max. and min. transmission | 0.765 5 and 0.748 2 | 0.768 9 and 0.750 1 |
| Data/restraints/parameters | 5 196/7/359 | 5 024/14/398 |
| Goodness-of-fit on F2 | 1.078 | 1.055 |
| Final R indices [I>2σ(I)] | R1=0.032 6, wR2=0.085 5 | R1=0.037 4, wR2=0.100 0 |
| R indices(all data) | R1=0.034 3, wR2=0.086 7 | R1=0.049 2, wR2=0.106 5 |
| (Δρ)max/(e·nm-3) | 1 791 | 1 194 |
| (Δρ)min/(e·nm-3) | -747 | -735 |
 下载: 导出CSV
下载: 导出CSV
表 2 配合物的部分键长和键角
Table 2. Parts of bond lengths(nm) and bond angles(°)
| C1 | |||||
| Sn(1)—C(13) | 0.214 4(3) | Sn(1)—C(20) | 0.215 1(3) | Sn(1)—O(1) | 0.215 9(2) |
| Sn(1)—N(1) | 0.221 9(2) | Sn(1)—O(2) | 0.230 6(2) | Sn(1)—O(5) | 0.254 8(3) |
| Sn(1)—O(2)i | 0.264 56(19) | ||||
| C(13)—Sn(1)—C(20) | 158.39(13) | C(13)—Sn(1)—O(1) | 93.92(11) | C(20)—Sn(1)—O(1) | 94.26(11) |
| C(13)—Sn(1)—N(1) | 102.62(11) | C(20)—Sn(1)—N(1) | 98.95(11) | O(1)—Sn(1)—N(1) | 70.90(8) |
| C(13)—Sn(1)—O(2) | 92.01(11) | C(20)—Sn(1)—O(2) | 94.03(11) | O(1)—Sn(1)—O(2) | 141.30(8) |
| N(1)—Sn(1)—O(2) | 70.49(8) | C(13)—Sn(1)—O(5) | 80.33(14) | C(20)—Sn(1)—O(5) | 82.79(14) |
| O(1)—Sn(1)—O(5) | 73.92(9) | N(1)—Sn(1)—O(5) | 144.81(10) | O(2)—Sn(1)—O(5) | 144.69(9) |
| C(13)—Sn(1)—O(2)i | 80.850(107) | C(20)—Sn(1)—O(2)i | 82.810(102) | O(2)—Sn(1)—O(2)i | 65.606(71) |
| O(5)—Sn(1)—O(2)i | 79.134(92) | ||||
| C2 | |||||
| Sn(1)—C(17) | 0.213 7(4) | Sn(1)—C(10) | 0.214 0(4) | Sn(1)—O(1) | 0.215 3(3) |
| Sn(1)—N(1) | 0.222 4(3) | Sn(1)—O(2) | 0.230 7(3) | Sn(1)—O(4) | 0.253 3(4) |
| Sn(1)—O(2)i | 0.271 04(27) | ||||
| C(17)—Sn(1)—C(10) | 158.05(17) | C(17)—Sn(1)—O(1) | 94.22(15) | C(10)—Sn(1)—O(1) | 96.04(14) |
| C(17)—Sn(1)—N(1) | 102.18(15) | C(10)—Sn(1)—N(1) | 99.50(15) | O(1)—Sn(1)—N(1) | 71.08(11) |
| C(17)—Sn(1)—O(2) | 92.15(14) | C(10)—Sn(1)—O(2) | 91.82(14) | O(1)—Sn(1)—O(2) | 141.52(10) |
| N(1)—Sn(1)—O(2) | 70.47(11) | C(17)—Sn(1)—O(4) | 83.65(17) | C(10)—Sn(1)—O(4) | 80.19(17) |
| O(1)—Sn(1)—O(4) | 75.61(12) | N(1)—Sn(1)—O(4) | 146.49(13) | O(2)—Sn(1)—O(4) | 142.86(12) |
| C(10)—Sn(1)—O(2)i | 81.279(142) | C(17)—Sn(1)—O(2)i | 80.940(132) | O(2)—Sn(1)—O(2)i | 64.714(87) |
| O(4)—Sn(1)—O(2)i | 78.222(102) | ||||
| Symmetry code for complex C1:i 2-x, 2-y, 1-z; C2:i1-x, 2-y, -z. | |||||
下载: 导出CSV
表 3 配合物对癌细胞的体外抑制活性
Table 3. Inhibition activiities of complexes against cancer cell in vitro
| IC50/(μmol·L-1) | |||
| NCI-H460 | HepG2 | MCF7 | |
| C1 | 5.34±0.37 | 3.00±0.11 | 4.57±0.12 |
| C2 | 8.41±0.25 | 7.27±0.34 | 5.20±0.18 |
| Carboplatin | 7.26±0.32 | 7.70±0.25 | 8.22±0.41 |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们


 下载:
下载: