Citation:
Ruijing Wang, Shixuan Wang, Chenghui Zhu, Jianze Zhang, Richard N. Zare, Chongqin Zhu, Xinxing Zhang. Increased acidity of nitrogen heterocyclic compounds on water microdroplets facilitates CO2 capture[J]. Chinese Chemical Letters,
2026, 37(4): 111547.
doi:
10.1016/j.cclet.2025.111547
Increased acidity of nitrogen heterocyclic compounds on water microdroplets facilitates CO2 capture
English
Increased acidity of nitrogen heterocyclic compounds on water microdroplets facilitates CO2 capture
College of Chemistry, Frontiers Science Center for New Organic Matter, State Key Laboratory of Advanced Chemical Power Sources, Key Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), Tianjin Key Laboratory of Biosensing and Molecular Recognition, Renewable Energy Conversion and Storage Center (ReCAST), Nankai University, Tianjin 300071, China
b.
Key Laboratory of Theoretical and Computational Photochemistry, Ministry of Education, College of Chemistry, Beijing Normal University, Beijing 100875, China
c.
Department of Chemistry, Stanford University, Stanford, CA 94305, United States
Received Date:
23 February 2025 Accepted Date:
04 July 2025 Revised Date:
06 May 2025 Available Online:
15 April 2026
Abstract:
Species at the air-water interface of microdroplets often display distinct acidity compared to the bulk. In this study, we report that pyrrole, imidazole, pyrazole, and 2H-1,2,3-triazole, a group of five-membered, planar, aromatic, nitrogen heterocyclic compounds that are basic in bulk water, exhibit strong acidity on microdroplets. The deprotonated anions of pyrrole, imidazole, and pyrazole can further react with CO2 to generate the corresponding carboxylic acids, but the triazole anion does not react with CO2. Calculation shows that partial solvation and the electric field on the air-water interface of the microdroplets are the main causes for the increased acidity, and the unique solvation structure of the triazole anion at the interface causes the reactive sites to be shielded by interfacial water molecules, thereby hindering reaction with CO2. These results demonstrate that the electric field and solvation structure of ions at the air-water interface play a decisive role in microdroplet chemistry for these compounds. We anticipate that the unique acidity and reactivity on microdroplets provide a new avenue that is rich in opportunities for green chemistry.
Excessive CO2 emissions are leading to significant environmental changes, including global warming and ocean acidification [1,2]. CO2 capture and sustainable conversion are considered as promising approaches to address this issue [3–6]. Current methods for capturing, transforming, and utilizing CO2 include physical or chemical adsorption [6], electrocatalytic reduction [7], photocatalytic reduction [8], and biological transformation [9]. Among these, amine-based compounds are widely employed as chemical adsorbents because they can employ the lone pairs of electrons on nitrogen to form N—C bonds with CO2 [10]. Monoethanolamine (HO—CH2—CH2—NH2) is extensively used in industrial CO2 removal due to its superior CO2 adsorption capabilities. However, regeneration of CO2 from it requires substantial energy input and is associated with challenges such as corrosion, high volatility, and limited thermal stability [11–15]. Attempts have been made to address these drawbacks by integrating amines with materials such as metal-organic frameworks [16] and ionic liquids [17], but some amine-derived, task-specific ionic liquid adsorbents still exhibit sluggish CO2 capture kinetics caused by intermolecular hydrogen bonding [18,19]. To mitigate this issue, Gurkan et al. introduced aprotic heterocyclic anion-containing ionic liquids that reduce the hydrogen-bonding network [20]. These anions, featuring deprotonated tertiary amine ring structures, exhibit rapid CO2 absorption rates and maintain low viscosity both before and after reaction with CO2 [21]. Nevertheless, challenges such as complex purification processes [22], potential toxicity concerns [23], and insufficient long-term environmental impact studies [24] remain. Therefore, the development of a strategy that enables efficient, simple, and sustainable CO2 capture and conversion would represent a significant advance.
Water microdroplets have garnered significant attention due to their unique properties [25–37]. Compared to bulk water, microdroplets can accelerate reaction rates by several orders of magnitude and facilitate reactions that are otherwise unattainable in bulk water. The air-water interface of water microdroplets plays an important role in the chemical reactions [30]: it has a high electric field in the range of 109 V/m [25,26], and the pH value of water and acidity of compounds at the air-water interface appear to be highly different from the bulk. Both theory and experiment revealed that the water surface was acidic compared to the bulk [38–41], but controversy remains [42]. Specifically, for water microdroplets, Raman microscopy with pH probes suggested the accumulation of protons at the air-water interface of microdroplets, so that the surface was more acidic than the center of the microdroplet [43]. Further, protonation and deprotonation experiments at the outer surface of microdroplets [44,45] showed that water was Brønsted neutral at pH 3–4 (rather than at pH 7 as bulk water), indicating that interfacial water may be more dissociated than in the bulk. Similarly, a recent theoretical study showed that both OH- and H3O+ prefer to accumulate at air-water interface and form an electric double layer, with H3O+ preferentially residing in the topmost layer, but OH- being enriched in the deeper interfacial layer [46]. Taking advantage of the unique pH and acidity of microdroplets, several studies have succeeded in the accelerated synthesis of organic compounds [47–49].
Here, we focus on the unique acidity of several different five-membered nitrogen heterocyclic compounds: pyrrole (pyrr), imidazole (imi), pyrazole (pyra), and 2H-1,2,3-triazole (tz), at the microdroplet surface. Intriguingly, although these compounds are inherently basic in bulk water, the unique interfacial properties of microdroplets facilitate their deprotonation, leading to the formation of conjugated anions. The anions of pyrr, imi, and pyra can react with CO2 to form the corresponding carboxylic acids, but the tz anion does not react with CO2. Through a combination of experiments and simulations, we found that the increased acidity was a result of partial solvation and the electric field at the interface of the microdroplets, and the reactions between these anions and CO2 are highly dependent on their partial solvation structures at the air-water interface. Even though the tz anion exhibited the highest intensity in the experiments, its reactive sites were shielded by the interfacial water molecules, hindering the reaction with CO2. Previously, microdroplet chemistry has been successfully utilized for CO2 capture [50–56], but here we achieved this goal by taking advantage of the unique acidity at the interface of the microdroplets.
To validate the unique acidity and CO2 reactivity of these five-membered nitrogen heterocycles at the microdroplet interface, we combined experimental and computational approaches. The aqueous solutions of pyrr, imi, pyra, and tz were injected into a fused silica capillary at a flow rate of 15 µL/min using a syringe pump. High-pressure N2 sheath gas (80 psi) was introduced through a coaxial capillary to generate microdroplets, and the resulting spray directly entered the inlet of the mass spectrometer. For CO2 capture experiments, the sheath gas was switched from N2 to CO2 under the same pressure conditions. To elucidate the reaction mechanisms, a series of computational investigations were carried out. We first used classical molecular dynamics (MD) and ab initio MD (AIMD) coupled with advanced free energy calculations to study the solvation and reactive uptake of pyrr, imi, pyra, and tz molecules at the air-water interface. Subsequently, AIMD simulations combined with the stepwise multisubphase space metadynamics (SMS-MetaD) method [57] were conducted to explore the free energy profiles of deprotonation. Finally, classical MD simulations were employed to investigate the solvation structures of these four anions at the air-water interface to further explore their different reactivity with CO2. Detailed methods are provided in Supporting information.
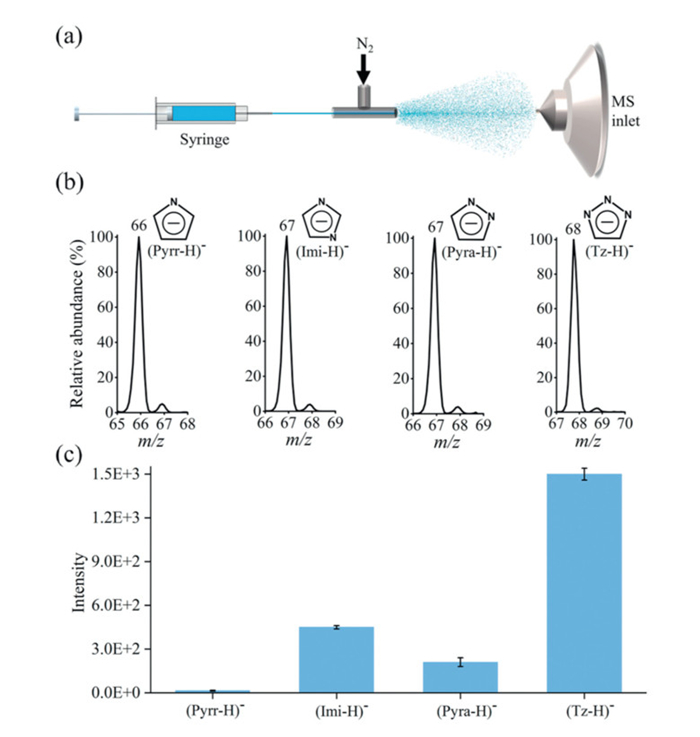
Fig. 1a illustrates the experimental setup, the distance between the capillary end and the mass spectrometer inlet was defined as the microdroplet flight (reaction) distance. Fig. 1b presents the typical mass spectra of deprotonated anions produced by spraying aqueous solutions of pyrr (m/z 66), imi (m/z 67), pyra (m/z 67), and tz (m/z 68) in air. The hydrogen on the N—H bonds of these four molecules are slightly acidic in water solutions. The pKa values of pyrr, imi, pyra, and tz are 17.5 [58], 14.5 [59], 14.2 [60], and 9.4 [61], respectively. Note that the pKa of water is 14 [59], suggesting that pyrr, imi, and pyra are much weaker acids than water in aqueous solutions, and tz is a stronger acid than water, but much weaker than formic acid (pKa 3.8). However, in microdroplets, all four molecules can be easily deprotonated, showing strong anionic signals in the mass spectra. Fig. 1c displays the relative deprotonation ability, i.e., the relative anionic mass spectral signal strength with 1 mmol/L aqueous solutions under the same experimental conditions. Fig. 1c indicates that tz has the strongest deprotonation ability, followed by imi, pyra, and finally pyrr, which generally aligns with the acidity order of these four compounds in aqueous solutions.
Figure 1
Figure 1.
Deprotonation of pyrr, imi, pyra, and tz in water microdroplets. (a) Experimental setup. (b) Negative mode mass spectra showing the deprotonated anions of pyrr, imi, pyra, and tz at a reaction distance of 10 mm and a sheath gas pressure of 80 psi. (c) Comparison of the mass spectral intensities of the deprotonated pyrr, imi, pyra, and tz anions under identical experimental conditions.
To explore the mechanisms underlying the increased acidity, we first investigated the solvation and reactive uptake of pyrr, pyra, imi, and tz molecules at the air-water interface using classical MD and AIMD coupled with advanced free energy calculations. Classical MD simulations in combination with umbrella sampling techniques were used to determine the free energy profiles of these molecules transitioning from the bulk phase into the gas phase through the air-water interface (Fig. S1a in Supporting information). Figs. S1b and c (Supporting information) show the density profile of water along the Z-axis and the free energy profile of a pyrr molecule (C4H5N) moving from the bulk water phase to the gas phase, respectively. Fig. S1c reveals a global minimum at the air-water interface, suggesting that pyrr molecules prefer to stay at the air-water interface. Similarly, pyra, imi, and tz molecules also prefer to reside at the air-water interface (Fig. S2 in Supporting information). Note that the global minimum relative to the bulk water represents the interfacial preference free energy, ΔFp, calculated to range between 1.5 kcal/mol and 2.5 kcal/mol. This indicates that five-membered nitrogen heterocycle molecules have an affinity for the air-water interface, promoting their further deprotonation.
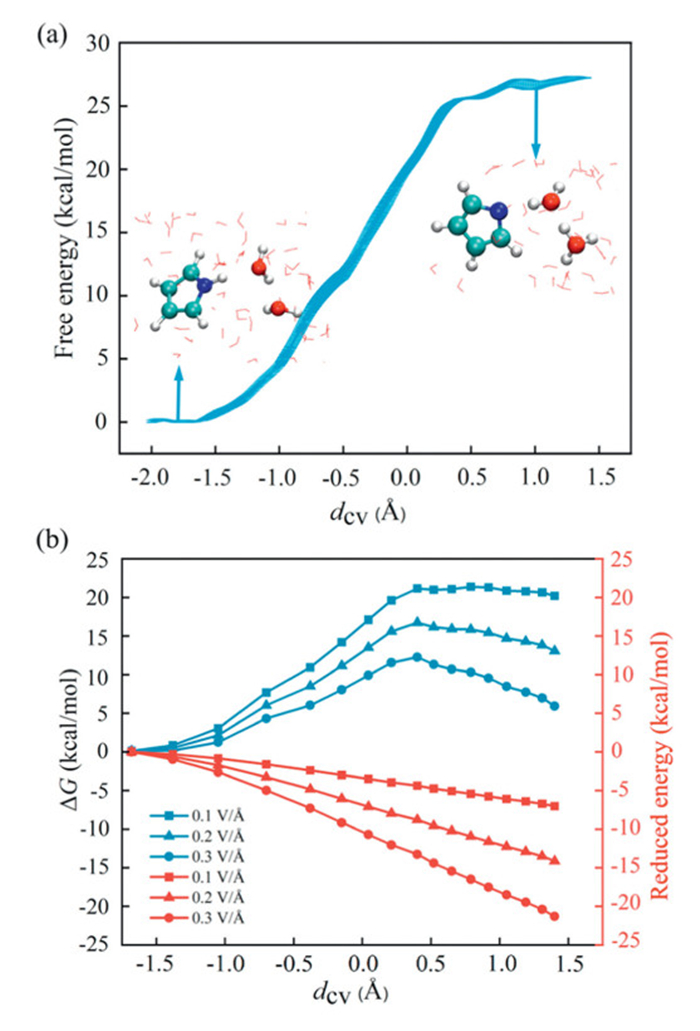
Next, we conducted AIMD simulations coupled with the SMS-MetaD method to investigate the free energy profiles of the deprotonation of these four molecules at the air-water interface. By linearly combining the formation and breaking of chemical bonds involved in the reaction, we constructed collective variables (CVs) (Fig. S3 in Supporting information), which effectively distinguish reactants, transition states, and products [62–64]. Fig. 2a shows the free energy profile of the dissociation of a pyrr molecule at the air-water interface. A minimum is observed at dcv = −1.8 Å, corresponding to the undissociated pyrr molecule, while for dcv > 0.75 Å, the free energy profile flattens, corresponding to the C4H4N- and H5O2+ ion pairs. The free energy difference is the dissociation free energy, ΔGd, which is ~27.3 kcal/mol at the air-water interface. In contrast, the ΔGd of C4H5N in the gas phase is as high as 57.37 kcal/mol (Fig. S4 in Supporting information). Clearly, although the partial solvation effect at the air-water interface significantly reduces the ΔGd for pyrr, its value remains relatively high, indicating that the partial solvation effect alone at the air-water interface is insufficient to drive the dissociation of the pyrr molecule. Therefore, we further investigated the effect of the electric field (EF) at the air-water interface on the dissociation of the pyrr molecule. Specifically, the EF was applied to the structures during the reaction along the direction of the molecular dipole moment. Fig. 2b illustrates the free energy profiles for the dissociation of C4H5N + 2H2O into C4H5N- and H5O2+ under various EF strengths. As the EF increases, the free energy of dissociation decreases. At EF strengths of 0.1, 0.2, and 0.3 V/Å, the dissociation free energy decreased by 6.9, 10.5, and 15.0 kcal/mol, respectively, with corresponding ΔGd values of 20.4, 16.8, and 12.3 kcal/mol. This indicates that the combined effect of the high electric field and partial solvation of the molecule at the air-water interface is what promotes the dissociation of C4H5N.
Figure 2
Figure 2.
(a) Free energy profile for the dissociation of pyrr at the air-water interface. (b) Dissociation free energy (ΔG) profiles (blue lines) and the corresponding energy reduction profiles (red lines) of pyrr at the air-water interface under EF strengths of 0.1, 0.2, and 0.3 V/Å.
In addition to the pyrr molecule, the combined effect of the high EF and partial solvation at the air-water interface can also promote the dissociation of pyra, imi, and tz. Specifically, at an EF strength of E = 0.3 V/Å, the ΔGd values for pyra, imi, and tz molecules are 12.2, 11.7, and 8.0 kcal/mol, respectively (Figs. S5-S7 in Supporting information). In summary, the deprotonation abilities of pyrr, imi, pyra, and tz follow the trend tz > imi > pyra > pyrr at the air-water interface, which is in good agreement with the experimental results (Fig. 1c).
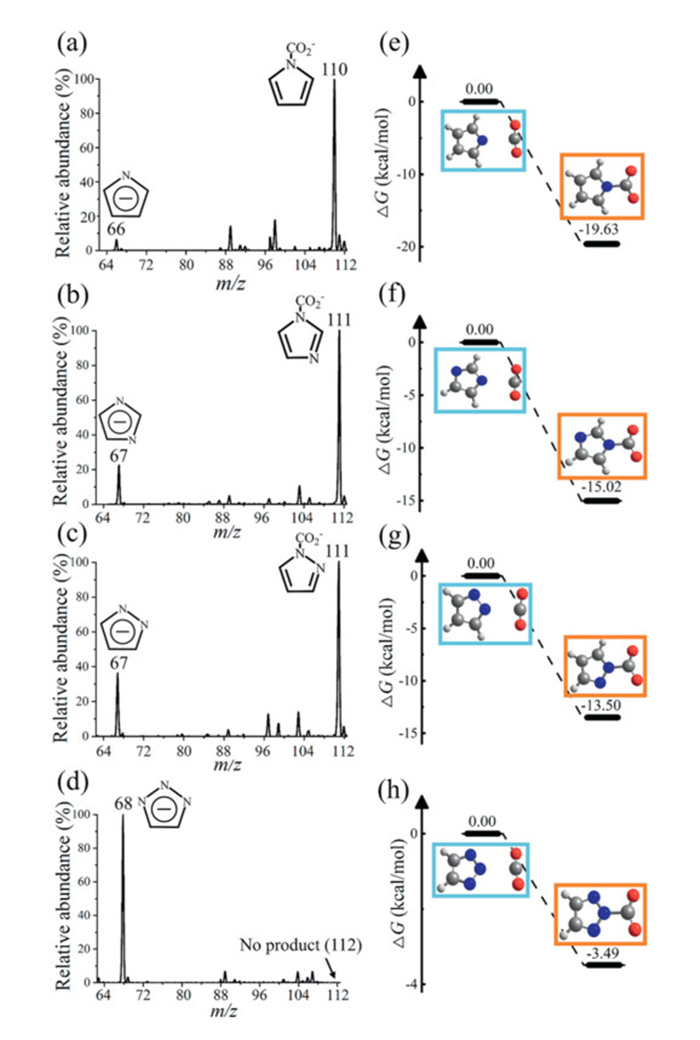
We next employed CO2 as the sheath gas to study how the four deprotonated five-membered nitrogen heterocyclic anions form C—N bonds with CO2. The mass spectra are presented in Fig. 3. Pyrr, imi, and pyra exhibited new peaks at m/z = 110 or m/z = 111 (Figs. 3a-c), indicating that the reactions with CO2 form the corresponding carboxylic acids. However, the tz anion does not react with CO2 (Fig. 3d). The yields are calculated as ∑I(product)/∑[I(product) + I(reactant)]. The pyrr anion exhibits the highest yield of 93% (Fig. 3a), followed by the imi anion at 83% (Fig. 3b), and the pyra anion at 71% (Fig. 3c). Collision-induced dissociation (CID) spectra provide structural information for these products (Fig. S8 in Supporting information). Quantum chemistry calculations support the experimental observation that the anions of pyrr, imi, and pyra capture CO2. As shown in Fig. 3, the reactions between these anions and CO2 are barrier-free. However, we observed no products for tz, which was initially puzzling.
Figure 3
Figure 3.
Typical mass spectra showing the reactions of (a) pyrr, (b) imi, (c) pyra, and (d) tz anions with CO2 on microdroplets and energy profiles for the reactions of (e) pyrr, (f) imi, (g) pyra, and (h) tz anions with CO2. Relative energies (in kcal/mol) of reactant complexes and product complexes were calculated at the CCSD(T)/cc-pVTZ//B3LYP/aug-cc-pVDZ level of theory.
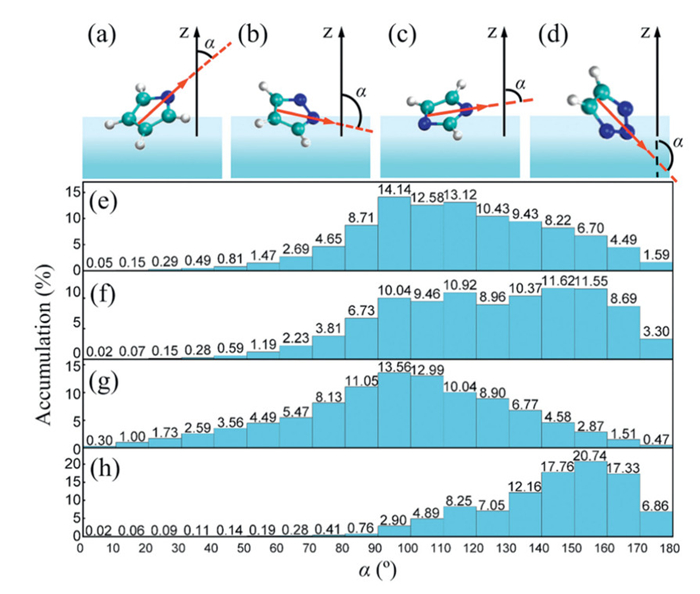
To further investigate the different reactivity with CO2 of these four anions, we employed classical MD simulations to study their solvation structures at the air-water interface (detailed methods are provided in Supporting information). We analyzed the distribution of the angle α for these anions at the interface, where α is defined as the angle between the Z-axis and the vector connecting the midpoint of the bond opposite the nitrogen atom and pointing toward the nitrogen atom (Figs. 4a-d). Figs. 4e-h present the distribution probability of α. An angle α less than 90 degrees indicates that the reactive site of the anion is not shielded by water molecules, whereas an angle greater than 90 degrees indicates that the reactive site is shielded. It is evident that, except for the tz anion, the other three anions exhibit a probability greater than 15% for α < 90°, allowing them enough chance to collide with gaseous CO2. In contrast, the tz anion shows a probability of less than 2% for α < 90°, indicating its tendency to be buried in the surrounding water molecules as illustrated in Fig. 4h. This explains why no product was observed for tz—the reactive sites of the tz anion were shielded by water, preventing efficient reaction with CO2.
Figure 4
Figure 4.
Typical configurations of (a) pyrr, (b) pyra, (c) imi, and (d) tz anions at the air-water interface. Distribution of the α angles for (e) pyrr, (f) pyra, (g) imi, and (h) tz anions at the air-water interface.
There is another possibility for the observation that the tz anion does not react with CO2. The negative charge is shared by the three N atoms on the tz anion [65], so the basicity, or the ability of donating the negative charge by the tz anion to CO2 should be lower compared to the other three anions. As a result, the reaction between tz anion and CO2 yields the least energy in all the four cases (Fig. 3). These facts might also contribute to the inertness of the tz anion towards the reaction with CO2.
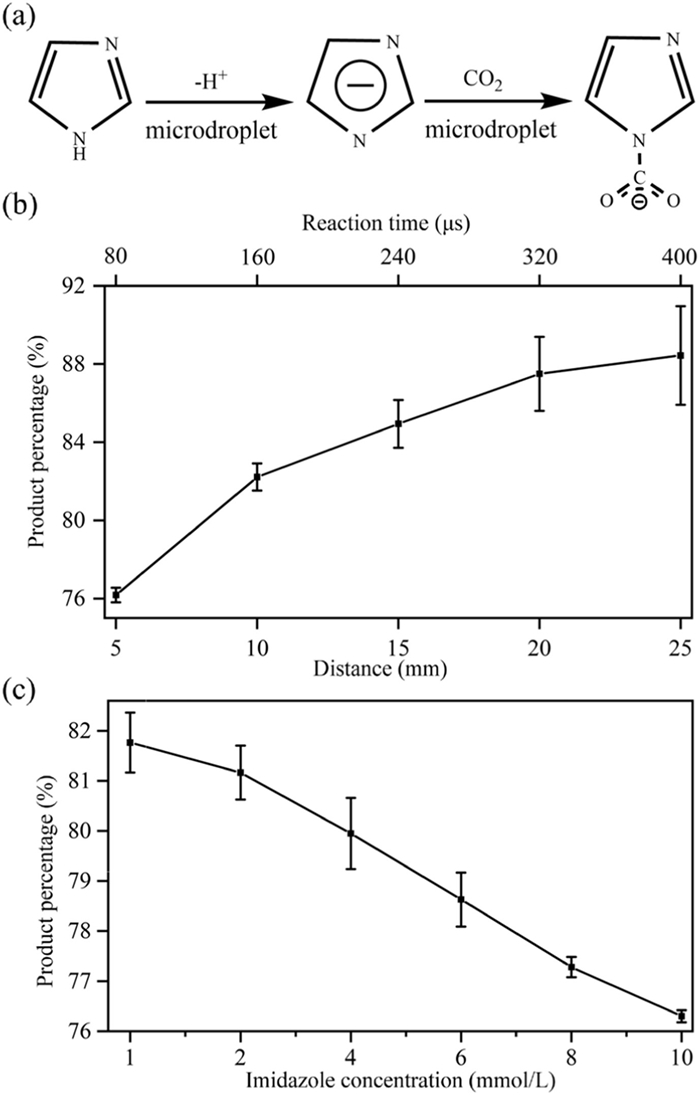
Finally, we used imi as an example to show the reaction mechanism of CO2 capture by a five-membered nitrogen heterocycle at the microdroplet interface (Fig. 5a). The imi molecule first deprotonates at the interface, then engages in nucleophilic attack on CO2, facilitating the transfer of a negative charge from the imi ring to CO2. Pyra and pyrr follow similar mechanisms. By varying the reaction distance from 5 mm to 25 mm (i.e., adjusting the reaction time from 80 µs to 400 µs [27]), the product percentage increases significantly (Fig. 5b), indicating that the reaction indeed occurs on the microdroplets in the air, rather than in the gas phase inside the mass spectrometer. We further studied the effect of reactant concentration on the product percentage (Fig. 5c) and found that the product percentage was higher at lower concentrations. This was because lower concentrations render higher fractions of the molecules at the microdroplet surface, which aligned with previous studies [66].
Figure 5
Figure 5.
Reactions between imi and CO2 in water microdroplets. (a) The reaction mechanism. (b) Product percentage as a function of reaction distance at a concentration of 1 mmol/L by adjusting the distance between the nebulizer and the mass spectrometer inlet. (c) Product percentage as a function of imi concentration when the reaction distance is 10 mm and the CO2 sheath gas pressure is 80 psi.
In summary, we systematically investigated the deprotonation of four five-membered nitrogen-containing heterocyclic compounds on water microdroplets, all of which deprotonated easily at the microdroplet air-water interface to form anions. Except for tz, the deprotonated anions can further react with CO2 to form the corresponding carboxylic acids. Importantly, our simulations and experimental results demonstrate that the electric field and partial solvation are the main reasons for the increased acidity of these compounds. The solvation structure of anions at the interface plays a decisive role in the reactions with CO2 occurring at the air-water interface. Although the tz anion exhibits the highest concentration at the interface and its reaction with CO2 is barrier-free, its unique interfacial solvation structure causes the reactive sites to be shielded by interfacial water molecules, thereby hindering efficient reaction with CO2. We anticipate that the unique acidity and reactivity on microdroplets provides a new avenue that is rich in opportunities for green chemistry.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statement
Ruijing Wang: Investigation, Data curation. Shixuan Wang: Investigation, Data curation. Chenghui Zhu: Investigation. Jianze Zhang: Investigation. Richard N. Zare: Writing – original draft, Supervision, Investigation. Chongqin Zhu: Software, Investigation. Xinxing Zhang: Supervision, Conceptualization, Writing – original draft.
Acknowledgments
X. Zhang acknowledges the National Key R&D Program of China (Nos. 2023YFE0124200 and 2023YFA1507203), the National Natural Science Foundation of China (No. 22325402), the Haihe Laboratory of Sustainable Chemical Transformations (No. 25HHWCSS00018), and the Frontiers Science Center for New Organic Matter at Nankai University. C.Z. was supported by the National Key Research and Development Program of China (Nos. 2024YFA1509600, 2021YFA1500700), the Beijing Natural Science Foundation (No. JQ24005), the National Natural Science Foundation of China (No. 22173011), the Training Program of the Major Research Plan of the National Natural Science Foundation of China (No. 92477140) and "the Fundamental Research Funds for the Central Universities". R.N.Z. acknowledges the US Air Force Office of Scientific Research through the Multidisciplinary University Research Initiative (MURI) program (No. AFOSR FA9550-21-1-0170).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.111547.
K. Gong, Y. Meng, R.N. Zare, J. Xie, J. Am. Chem. Soc. 146 (2024) 8576–8584. doi: 10.1021/jacs.4c00529
[57]
Y.G. Fang, X. Li, Y. Gao, et al., J. Chem. Phys. 157 (2022) 214111. doi: 10.1063/5.0098269
[58]
A.R. Katritzky, C.W. Rees, Comprehensive heterocyclic chemistry, in: C.W. Bird, G.W.H. Cheeseman, (Eds.), Five-membered Rings with One Oxygen, Sulfur or Nitrogen Atom., New York, 1984.
[59]
H. Walba, R.W. Isensee, J. Org. Chem. 26 (1961) 2789–2791. doi: 10.1021/jo01066a039
[60]
M.J. Alam, O. Alam, P. Alam, et al., Int. J. Pharm. Sci. Res. 6 (2015) 1433–1442.
K.R. Wilson, A.M. Prophet, G. Rovelli, et al., Chem. Sci. 11 (2020) 8533–8545. doi: 10.1039/d0sc03189e
Figure 1
Deprotonation of pyrr, imi, pyra, and tz in water microdroplets. (a) Experimental setup. (b) Negative mode mass spectra showing the deprotonated anions of pyrr, imi, pyra, and tz at a reaction distance of 10 mm and a sheath gas pressure of 80 psi. (c) Comparison of the mass spectral intensities of the deprotonated pyrr, imi, pyra, and tz anions under identical experimental conditions.
Figure 2
(a) Free energy profile for the dissociation of pyrr at the air-water interface. (b) Dissociation free energy (ΔG) profiles (blue lines) and the corresponding energy reduction profiles (red lines) of pyrr at the air-water interface under EF strengths of 0.1, 0.2, and 0.3 V/Å.
Figure 3
Typical mass spectra showing the reactions of (a) pyrr, (b) imi, (c) pyra, and (d) tz anions with CO2 on microdroplets and energy profiles for the reactions of (e) pyrr, (f) imi, (g) pyra, and (h) tz anions with CO2. Relative energies (in kcal/mol) of reactant complexes and product complexes were calculated at the CCSD(T)/cc-pVTZ//B3LYP/aug-cc-pVDZ level of theory.
Figure 4
Typical configurations of (a) pyrr, (b) pyra, (c) imi, and (d) tz anions at the air-water interface. Distribution of the α angles for (e) pyrr, (f) pyra, (g) imi, and (h) tz anions at the air-water interface.
Figure 5
Reactions between imi and CO2 in water microdroplets. (a) The reaction mechanism. (b) Product percentage as a function of reaction distance at a concentration of 1 mmol/L by adjusting the distance between the nebulizer and the mass spectrometer inlet. (c) Product percentage as a function of imi concentration when the reaction distance is 10 mm and the CO2 sheath gas pressure is 80 psi.

 DownLoad:
DownLoad:

 下载:
下载:






 下载:
下载:
