
Scheme 1.
Synthesis and application of the designed arylene-bridged bis(benzimidazolium).
Fine-tuning redox ability of arylene-bridged bis(benzimidazolium) for electrochromism and visible-light photocatalysis
Jing Wang , Zenghui Li , Xiaoyang Liu , Bochao Su , Honghong Gong , Chao Feng , Guoping Li , Gang He , Bin Rao

Organic multistage redox systems (≥ two electrons), which involve electron transfer processes, have significantly contributed to the utilization of organic materials in various applications such as functional dyes, electronic devices, photovoltaic batteries, data storage, and organic field-effect transistors [1–5]. These redox molecules typically consist of two end groups X, and a π-system linkage, allowing them to undergo three redox states through the process of reduction and oxidation (Scheme 1a) [6]. The representative redox molecules, including benzoquinone (Ⅰ), tetraaza-substituted olefins (Ⅱ) and tetrathiafulvalene (Ⅲ) and derivatives have been well developed for their remarkable redox behavior [7–10]. Additionally, viologens (Ⅳ), which are built with two pyridinium end groups as electron acceptors, exhibit excellent redox ability through a two-step reversible one-electron reduction. As a result, viologens find wide applications in electrochromic display devices, photochromism, and other fields [11–21]. In these molecules, the presence of stable neutral or ionic radicals throughout the electron transfer process, known as multi-redox systems, determines the range and potential applications. Consequently, it is highly desirable for the scientific community to explore and design more sophisticated and controllable redox systems.
Herein, imidazolium was chosen as the end group to construct multistage redox molecules in combination with various π-system linkages. Imidazolium compounds serve as the derivatives of N-heterocyclic carbenes (NHCs), which possess excellent σ-electron-donating properties to stabilize transition metals and main group elements, as well as organic radical intermediates [22–25]. Recently, there has been a growing interest in the development of NHCs-based redox molecules. For instance, cyclic (alkyl)(amino)carbenes designed by the group of Bertrand [26,27], have been used to build up a range of redox molecules by incorporating various π-linkers (V), such as alkyne [28–30], olefin [31,32] and arene units [33,34]. Because of the exciting redox ability, they have been successfully applied in the field of redox flow batteries [35–37] and singlet fission materials [38,39]. Subsequently, the choice of cyclic diamino carbenes as the end groups to build redox systems (VI) was also achieved by the group of Ghadwal, isolating a series of stable redox intermediates [40–43]. However, the broader application of imidazolium-based redox molecules has yet to be fully explored. One possible reason for this is the relatively high redox potentials and the limited control over the available oxidation states.
To fine tune the redox ability, 6 different linkers including anthracene, multi-benzene, naphthalene and thiophene were investigated in combination with benzimidazolium units as ending groups, giving a series of arylene-bridged bis(benzimidazolium)triflates (ArBIm2+·2OTf, 1–62+·2[OTf–]). To briefly assess the redox properties of these compounds, a theoretical prediction of the HOMO/LUMO energies was conducted (Fig. S7 in Supporting information) [44]. The results indicated that the compound 12+, which contains an anthracene unit, is expected to exhibit the best performance in the redox process, thanks to its lowest energy gap [45,46]. As illustrated in Scheme 2, 1–62+·2[OTf–] bearing different π-linkers were successfully synthesized in a concise method. Starting from readily available N,N-diisopropylbenzene-1,2-diamine and corresponding aldehydes, ArBIm2+·2OTf precursors were prepared through a condensation reaction and subsequent H-atom abstraction in the presence of 2-bromoacetophenone according to the literature (Scheme S1 in Supporting information) [47]. Then anion exchange between bromide and triflate was achieved in the presence of methyl triflate at room temperature in the solvent of dichloromethane. Targeted 1–62+·2[OTf–] were obtained in one pot through three steps, giving satisfied yields in Scheme 2.
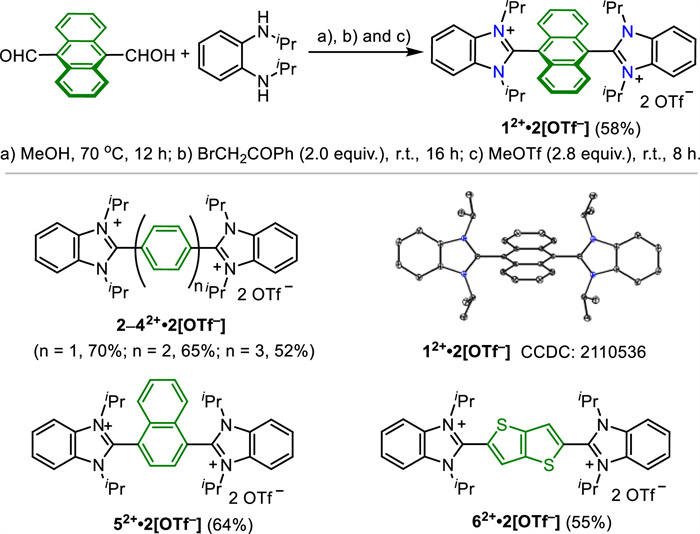
All these compounds exhibit good solubility in most polar solvents such as methanol, dimethyl sulfoxide, acetonitrile, and DMF. They were fully characterized by 1H NMR, 13C NMR, and HRMS measurement. Additionally, their stability was confirmed through thermogravimetric analysis (Fig. S2a in Supporting information). To gain insights into their solid-state structures, single crystals of compounds 12+·2[OTf–] and 42+·2[OTf–], were obtained by slow evaporation of mixed solvents, namely dichloromethane and methanol (Table S1 and Fig. S1 in Supporting information). In compound 12+·2[OTf–], due to the steric repulsion between the benzimidazolium units and the hydrogen atoms in the peri‑position of the anthracene core, the dihedral angle between the two planes is nearly orthogonal (88.36°). In compound 42+·2[OTf–], the dihedral angle between the benzimidazolium and benzene units is 74.12° (Fig. S1).
The photophysical properties of ArBIm2+·2OTf were investigated and the results are presented in Figs. 1a and b. Firstly, the UV−vis absorption properties of ArBIm2+·2OTf were examined in diluted DMF solutions, revealing strong absorption bands in the range of approximately 284–315 nm for 2–62+·2[OTf–]. Notably, the absorption spectrum of 12+·2[OTf–] exhibited a red-shift compared to other ArBIm2+·2OTf and showed three intense absorption bands at around 273, 381, and 403 nm. These absorption bands can be mainly attributed to the HOMO to LUMO transition (S0→S1, l = 419 nm, f = 0.22) and HOMO to LUMO+3 transition coupled with HOMO-5 to LUMO transition (S0→S11, l = 255 nm, f = 1.51), as indicated by TD-DFT calculations at B3LYP/6–311G(d, p) level theory (Figs. 1c and d). Furthermore, the photoluminescence (PL) spectra of ArBIm2+·2OTf were also measured in DMF, showing emission peaks ranging from 364 nm to 436 nm (Fig. 1b). It should be noted that 12+·2[OTf–] exhibited good absorption in the range of 480–430 nm, implying the potential application in visible-light photocatalysis [48,49].
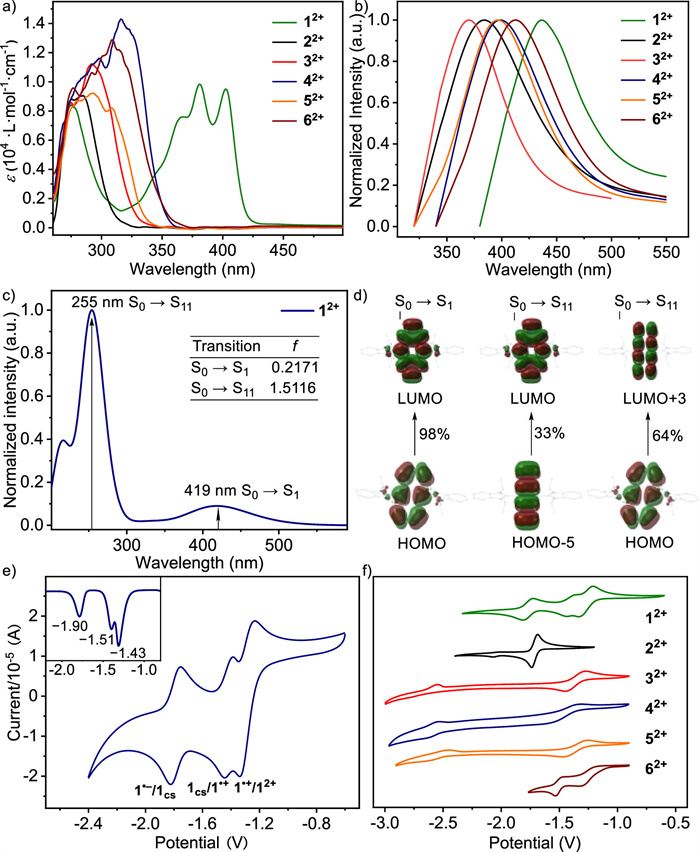
To investigate the redox properties of ArBIm2+·2OTf, cyclic voltammetry (CV) was performed in DMF (Figs. 1e and f). CV curve of 12+·2[OTf–] showed three reversible single-electron reductions (E1/2 = –1.20, –1.40 and –1.73 vs. Fc/Fc+), indicating that the combination of benzimidazolium units and anthracene motif would adjust the multistage redox system (> two-electron transfer) as we expected. This was further confirmed by differential pulse voltammetry (DPV) in Fig. 1e. Based on CV and DPV curves of 12+·2[OTf–], it can be inferred that three reduced species would be generated through the successive one-electron reductions. These species include a radical cation 1•+, a neutral species 1CS (close-shell singlet 1CS, open-shell singlet 1OS, and triplet 1T), and a radical anion 1•– (Schemes S2 and S4 in Supporting information). The CV curves of 2–52+·2[OTf–] respectively exhibited one reversible redox at 1E1/2 = –1.67, –1.29, –1.34 and –1.27 V vs. Fc/Fc+, and another irreversible wave at 2E1/2 = –2.01, –2.44, –2.51 and –2.49 V vs. Fc/Fc+ (Fig. 1f and Fig. S3 in Supporting information). Compound 62+·2[OTf–] bearing fused-thiophene unit, showed two quasi-reversible waves (1E1/2 = –1.09 V, 2E1/2 = –1.47 V). Additionally, when scanning speeds ranging from 0.01 V/s to 1 V/s were tested for ArBIm2+•2OTf, the reversibility of the waves increased (Fig. S3), further confirming the influence of electron transfer rate on the stability of the reduced species. Due to the combination of benzimidazolium and the large π-extended conjugation system of the anthracene motif, 12+·2[OTf–] displayed four accessible redox states, in contrast to other ArBIm2+·2OTf. It is worth noting that, to the best of our knowledge, no more than three redox states have been observed in previously reported arylene-bridged bisimidazolium molecules [40–42].
Next, our research focuses on the application of ArBIm2+·2OTf in the field of electrochromism, taking inspiration from our previous work on functionalized viologens with a multistage redox process [50–52]. To explore this, we fabricated electrochromic devices (ECDs) using 12+·2[OTf–] as the active component, DMF as the solvent, and indium tin oxide (ITO)-coated glass as the electrodes. In the ECDs based on 12+·2[OTf–], applying a voltage of –3.9 V resulted in blue color, while a higher voltage of –4.2 V induced a red color (Fig. 2a). Observations of the ECDs under –3.9 V revealed strong absorption bands at approximately 561, 608, and 671 nm, indicating the accumulation of the radical cation 1•+ (Fig. 2b). As the potential was adjusted to –4.2 V, a decrease in absorption at approximately 561, 608, and 671 nm was observed, accompanied by an increase in absorption at approximately 526 nm. These changes signify the consumption of the radical cation 1•+ and its conversion to the neutral species 1CS (Fig. 2c). These color transitions correlate with the formation of the radical cation 1•+ from 12+ after one-electron uptake, followed by the conversion to the neutral species 1CS through the second reduction.

However, when the applied potential was increased to –5.2 V, the color of the ECD changed to black, possibly due to the high instability of the radical anion 1•– within the device. This instability may induce the limited reversibility of the electrochromic device for 12+·2[OTf–]. To address this issue, future work will focus on enhancing the stability of the radical intermediates by the introduction of bulky groups.
Furthermore, the 32+·2[OTf–] or 42+·2[OTf–]-based ECDs would undergo an irreversible color change from their original colorless state to black when subjected to a high external voltage of –1.2 V. This behavior might be attributed to the decomposition of the corresponding reduced species of 32+·2[OTf–] and 42+·2[OTf–]. On the other hand, the ECDs constructed with 22+·2[OTf–], 52+·2[OTf–] and 62+·2[OTf–] displayed red (–3.9 V), blue-violet (–4.2 V), and deep blue color (–4.3 V) respectively, and more importantly they can also go back to the colorless state reversibly (Figs. S4–S6 in Supporting information). Additionally, spectroelectrochemical characterizations of fabricated ECD based on 22+·2[OTf–], 52+·2[OTf–] and 62+·2[OTf–] were carried out (Figs. S4–S6). These experiments further support the notion that arylene-bridged bis(benzimidazolium) triflates can serve as a promising new class of electrochromic molecules.
To gain insights into the reduced species 1•+, 1CS and 1•– from 12+, the chemical reduction was carried out. When treated 12+·2[OTf–] with 1 equiv. potassium graphite (KC8) at –78 ℃, the mixture became deep blue color. And the UV−vis absorption was located at ca. 560 and 660 nm, which is in agreement with the display of ECD based on 12+·2[OTf–], as well as the calculated result by TD-DFT (Fig. 3d and Fig. S11 in Supporting information). Moreover, the electron paramagnetic resonance (EPR) measurement provided conclusive evidence regarding the organic radical nature of 1•+ with a g-factor value of 2.0031 (Fig. 2e). Additionally, the hyperfine structure of the experimental EPR 1•+ was reproduced for the coupling of the unpaired electron with nitrogen atoms and corresponding hydrogen atoms on the anthracene unit. This finding was in agreement with the calculated distribution of spin density, which was predominantly located on the anthracene unit (Fig. 2g). Next, the reduction of 12+ with 2 equiv. KC8 exhibited purple color and the absorption band was located at ca. 520 nm in the UV−vis spectrum (Fig. 2d, Scheme S2 in Supporting information). The treatment of 12+ with three equivalents KC8 in THF at –78 ℃ gave a dark green solution, and the corresponding EPR spectrum showed a multiline pattern with g-factor values of 2.0031 (Fig. 2f). Radical anion 1•– needs to store at low temperatures under dark conditions, otherwise, it would gradually decompose at room temperature after 48 h. The spin density distribution of radical anion 1•– was mainly on the benzimidazolium moiety (Fig. 2h).
Meanwhile, the radical cation 1•+ and anion 1•– may be also defined as mixed-valence compounds for the donor-acceptor behavior in the open-shell system. So the UV–vis spectra were measured from 800 nm to 1600 nm (Fig. S2b), and there were no apparent absorption peaks, implying 1•+ and 1•– as classing I compound for the absence of intervalence charge transfer (IV-CT) bands [53]. Attempt to isolate reduced intermediates (1•+, 1CS and 1•–) for the single crystal analysis failed. And the possible reason is that the carbon radical center may be not effectively protected by the surrounding group, such as the mesityl or 2,6-diisopropyl phenyl group, as indicated in Ghadwal's and Hansmann's work [33–35,40–43,54]. Nonetheless, the stepwise redox process exhibited by 12+ through its electron-accepting behavior is similar to that of viologen compounds in the field of electrochromism. The unique electrochemical properties and the ease of modification of arylene-bridged bis(benzimidazolium)triflates hold great potential for applications as advanced redox materials, offering exciting opportunities for further development.
Furthermore, the use of synthetic organic photocatalysts has garnered significant attention due to their ability to facilitate complex molecular transformations using light as a natural energy source [48,49]. In this regard, we reasoned that ArBIm2+·2OTf could function as effective organic photocatalysts owing to their reversible redox capability and favorable photophysical properties [55,56]. To evaluate the photocatalytic properties of ArBIm2+·2OTf, the visible-light-induced cross-dehydrogenative coupling (CDC) reaction between N-phenyl tetrahydroisoquinoline and diphenylphosphine oxide was chosen as a model reaction to form carbon-phosphorus bond in Scheme 3. After careful investigation of the reaction condition (Table S2 in Supporting information), the CDC reaction proceeded smoothly with a catalytic amount of 1–62+·2[OTf–] in the presence of white light under air conditions. Among them, catalyst 12+·2[OTf–] exhibited the best catalytic performance, leading to the desired product in 89% isolated yield with the full conversion of 7 in less than 12 h, which may be attributed to the good visible-light absorption and redox ability for the π-linker of anthracene unit. Conversely, less than 10% yield was observed under dark conditions or without a catalyst. Then, the substrates with different alkoxyl groups can also successfully react with 7, giving products 10–12 with isolated yields of 82%, 91% and 65%, respectively. Meanwhile, N-aryl tetrahydroisoquinoline substrates with bromo and methoxyl groups can also give the coupling products (13-16) in good yield (77%~90%). Based on our experimental findings and relevant literature, we propose a plausible reaction mechanism (Scheme S3 in Supporting information) [49,57,58]. These results clearly demonstrate the efficacy of 1 as an excellent organic photocatalyst for C-P bond formation, in comparison to other photocatalysts such as Eosin Y [57] and ruthenium- or iridium-based complexes (Table S3 in Supporting information) [59,60].

In summary, we have presented a family of arylene-bridged bis(benzimidazolium)triflates 1–62+·2[OTf–], which exhibit varying redox abilities depending on the different arylene linkers employed. Notably, 12+·2[OTf–] featuring an anthracene unit as π-linker demonstrates excellent electron transfer in four redox states. Due to the excellent redox and optoelectronic properties, 12+·2[OTf–] was successfully used as an electrochromic molecule under applied voltage in ECD, and also as an excellent photocatalyst to construct carbon–phosphorus bond via visible-light-induced cross-dehydrogenative coupling reaction. These findings not only expand the utility of NHC-based multistage redox systems as electrochromic materials but also position them as promising candidates for organic photocatalysis applications.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by Natural Science Foundation of China (Nos. 22001200, 22175138, 21875180). We thank Dr. Chao Feng, and Chang Huang at Instrument Analysis Center of Xi'an Jiaotong University for their assistance with NMR, HRMS, and X-ray crystallographic measurements. We thank Lijing Ma at State Key Laboratory of Multiphase Flow in Power Engineering of Xi'an Jiaotong University for her assistance with EPR measurements. We also acknowledge the support by the high-performance computing center of Xi'an Jiaotong University. We thank Xiaoming Zeng (Sichuan university) and Yuanting Su (Soochow University) for their valuable suggestion on the manuscript.
Supplementary material associated with this article can be found, in the online version, at doi:
T. Nishinaga, Organic Redox Systems: Synthesis, Properties, and Applications, John Wiley & Sons, 2015.
R.J. Mortimer, Chem. Soc. Rev. 26 (1997) 147–156. doi: 10.1039/cs9972600147
D.G. Kwabi, Y. Ji, M.J. Aziz, Chem. Rev. 120 (2020) 6467–6489. doi: 10.1021/acs.chemrev.9b00599
M.J. Sun, H.J. Nie, J.N. Yao, Y.W. Zhong, Chin. Chem. Lett. 26 (2015) 649–652. doi: 10.1016/j.cclet.2015.03.032
J. Feng, J.Y. Shao, H.J. Nie, Z.L. Gong, Y.W. Zhong, Chin. Chem. Lett. 29 (2018) 385–389. doi: 10.1016/j.cclet.2017.11.014
K. Deuchert, S. Hünig, Angew. Chem. Int. Ed. 17 (1978) 875–886. doi: 10.1002/anie.197808753
J. Broggi, T. Terme, P. Vanelle, Angew. Chem. Int. Ed. 53 (2014) 384–413. doi: 10.1002/anie.201209060
J.A. Murphy, J. Org. Chem. 79 (2014) 3731–3746. doi: 10.1021/jo500071u
E. Doni, J.A. Murphy, Chem. Commun. 50 (2014) 6073–6087. doi: 10.1039/C3CC48969H
M. Bendikov, F. Wudl, D.F. Perepichka, Chem. Rev. 104 (2004) 4891–4946. doi: 10.1021/cr030666m
L. Striepe, T. Baumgartner, Chem. Eur. J. 23 (2017) 16924–16940. doi: 10.1002/chem.201703348
T. Janoschka, N. Martin, U. Martin, et al., Nature 527 (2015) 78–81. doi: 10.1038/nature15746
G. Das, T. Skorjanc, S.K. Sharma, et al., J. Am. Chem. Soc. 139 (2017) 9558–9565. doi: 10.1021/jacs.7b02836
J. Winsberg, T. Hagemann, T. Janoschka, M.D. Hager, U.S. Schubert, Angew. Chem. Int. Ed. 56 (2017) 686–711. doi: 10.1002/anie.201604925
M. Stolar, J. Borau-Garcia, M. Toonen, T. Baumgartner, J. Am. Chem. Soc. 137 (2015) 3366–3371. doi: 10.1021/ja513258j
C.R. Bridges, A.M. Borys, V.A. Beland, J.R. Gaffen, T. Baumgartner, Chem. Sci. 11 (2020) 10483–10487. doi: 10.1039/d0sc04183a
Y. Dai, Z. Xie, M. Bao, C. Liu, Y. Su, Chem. Sci. 14 (2023) 3548–3553. doi: 10.1039/d3sc00102d
S. Liu, M. Zhou, T. Ma, et al., Chin. Chem. Lett. 31 (2020) 1690–1693. doi: 10.1016/j.cclet.2019.11.033
X.H. Zhou, Y. Fan, W.X. Li, et al., Chin. Chem. Lett. 31 (2020) 1757–1767. doi: 10.1016/j.cclet.2019.12.039
Z.Y. Dong, D.W. Zhang, X.Z. Jiang, et al., Chin. Chem. Lett. 24 (2013) 688–690. doi: 10.1016/j.cclet.2013.04.048
H.J. Wang, W.W. Xing, Z.H. Yu, et al., Chin. Chem. Lett. 35 (2024) 109183. doi: 10.1016/j.cclet.2023.109183
M.N. Hopkinson, C. Richter, M. Schedler, F. Glorius, Nature 510 (2014) 485–496. doi: 10.1038/nature13384
C.D. Martin, M. Soleilhavoup, Chem. Sci. 4 (2013) 3020–3030. doi: 10.1039/c3sc51174j
A. Doddi, M. Peters, M. Tamm, Chem. Rev. 119 (2019) 6994–7112. doi: 10.1021/acs.chemrev.8b00791
A.A. Danopoulos, T. Simler, P. Braunstein, Chem. Rev. 119 (2019) 3730–3961. doi: 10.1021/acs.chemrev.8b00505
R. Jazzar, M. Soleilhavoup, G. Bertrand, Chem. Rev. 120 (2020) 4141–4168. doi: 10.1021/acs.chemrev.0c00043
M. Soleilhavoup, G. Bertrand, Acc. Chem. Res. 48 (2014) 256–266.
Y. Li, K.C. Mondal, P.P. Samuel, et al., Angew. Chem. Int. Ed. 53 (2014) 4168–4172. doi: 10.1002/anie.201310975
L. Jin, M. Melaimi, L.L. Liu, G. Bertrand, Org. Chem. Front. 1 (2014) 351–354. doi: 10.1039/C4QO00072B
B.M. Barry, R.G. Soper, J. Hurmalainen, et al., Angew. Chem. Int. Ed. 57 (2018) 749–754. doi: 10.1002/anie.201711031
D. Munz, J. Chu, M. Melaimi, G. Bertrand, Angew. Chem. Int. Ed. 55 (2016) 12886–12890. doi: 10.1002/anie.201607537
M.K. Nayak, J. Stubbe, N.I. Neuman, et al., Chem. Eur. J. 26 (2020) 4425–4431. doi: 10.1002/chem.202000255
M.M. Hansmann, M. Melaimi, G. Bertrand, J. Am. Chem. Soc. 140 (2018) 2206–2213. doi: 10.1021/jacs.7b11184
M.M. Hansmann, M. Melaimi, D. Munz, G. Bertrand, J. Am. Chem. Soc. 140 (2018) 2546–2554. doi: 10.1021/jacs.7b11183
P.W. Antoni, T. Bruckhoff, M.M. Hansmann, J. Am. Chem. Soc. 141 (2019) 9701–9711. doi: 10.1021/jacs.9b04249
G. Kwon, Y. Ko, Y. Kim, K. Kim, K. Kang, Acc. Chem. Res. 54 (2021) 4423–4433. doi: 10.1021/acs.accounts.1c00590
H. Song, E. Pietrasiak, E. Lee, Acc. Chem. Res. 55 (2022) 2213–2223. doi: 10.1021/acs.accounts.2c00222
T. Ullrich, P. Pinter, J. Messelberger, et al., Angew. Chem. Int. Ed. 59 (2020) 7906–7914. doi: 10.1002/anie.202001286
J. Messelberger, A. Grünwald, P. Pinter, M.M. Hansmann, D. Munz, Chem. Sci. 9 (2018) 6107–6117. doi: 10.1039/c8sc01999a
D. Rottschaefer, N.K.T. Ho, B. Neumann, et al., Angew. Chem. Int. Ed. 57 (2018) 5838–5842. doi: 10.1002/anie.201713346
D. Rottschaefer, B. Neumann, H.G. Stammler, D.M. Andrada, R.S. Ghadwal, Chem. Sci. 9 (2018) 4970–4976. doi: 10.1039/C8SC01209A
D. Rottschäfer, J. Busch, B. Neumann, et al., Chem. Eur. J. 24 (2018) 16537–16542. doi: 10.1002/chem.201804524
A. Merschel, D. Rottschäfer, B. Neumann, et al., Angew. Chem. Int. Ed. 62 (2023) e202215244. doi: 10.1002/anie.202215244
D.D. Méndez-Hernández, P. Tarakeshwar, D. Gust, et al., J. Mol. Model. 19 (2013) 2845–2848. doi: 10.1007/s00894-012-1694-7
J.E. Anthony, Chem. Rev. 106 (2006) 5028–5048. doi: 10.1021/cr050966z
M. Yoshizawa, J.K. Klosterman, Chem. Soc. Rev. 43 (2014) 1885–1898. doi: 10.1039/C3CS60315F
D.D. Tanner, J.J. Chen, J. Org. Chem. 54 (1989) 3842–3846. doi: 10.1021/jo00277a020
L. Shi, W. Xia, Chem. Soc. Rev. 41 (2012) 7687–7697. doi: 10.1039/c2cs35203f
N.A. Romero, D.A. Nicewicz, Chem. Rev. 116 (2016) 10075–10166. doi: 10.1021/acs.chemrev.6b00057
W. Ma, L. Xu, S. Zhang, et al., J. Am. Chem. Soc. 143 (2021) 1590–1597. doi: 10.1021/jacs.0c12015
B. He, S. Zhang, Y. Zhang, et al., J. Am. Chem. Soc. 144 (2022) 4422–4430. doi: 10.1021/jacs.1c11577
W. Ma, S. Zhang, L. Xu, et al., Chin. Chem. Lett. 34 (2023) 107958. doi: 10.1016/j.cclet.2022.107958
A. Heckmann, C. Lambert, Angew. Chem. Int. Ed. 51 (2012) 326–392. doi: 10.1002/anie.201100944
P.W. Antoni, C. Golz, M.M. Hansmann, Angew. Chem. Int. Ed. 61 (2022) e202203064. doi: 10.1002/anie.202203064
T. Kodama, M. Kubo, W. Shinji, K. Ohkubo, M. Tobisu, Chem. Sci. 11 (2020) 12109–12117. doi: 10.1039/d0sc03958f
R. Miyajima, Y. Ooe, T. Miura, et al., J. Am. Chem. Soc. 145 (2023) 10236–10248. doi: 10.1021/jacs.3c01264
D.P. Hari, B. König, Org. Lett. 13 (2011) 3852–3855. doi: 10.1021/ol201376v
H. Bartling, A. Eisenhofer, B. König, R.M. Gschwind, J. Am. Chem. Soc. 138 (2016) 11860–11871. doi: 10.1021/jacs.6b06658
M. Rueping, S. Zhu, R.M. Koenigs, Chem. Commun. 47 (2011) 8679–8681. doi: 10.1039/c1cc12907d
W.J. Yoo, S. Kobayashi, Green Chem. 16 (2014) 2438–2442. doi: 10.1039/C4GC00058G

Scheme 1 Synthesis and application of the designed arylene-bridged bis(benzimidazolium).

Scheme 2 Synthetic route to the designed arylene-bridged bis(benzimidazolium)triflates (1–62+·2[OTf–]).

Figure 1 (a) UV–vis spectra of ArBIm2+·2OTf in DMF, [c = 1 mmol/L]. (b) Emission spectra of ArBIm2+·2OTf in DMF at 298 K. (c) Calculated UV–vis absorption spectrum of 12+. (d) Frontier molecular orbitals of 12+ and associated electronic transitions. (e) Cyclic voltammograms of ArBIm2+·2OTf [c = 1 mmol/L] performed in DMF with Bu4NPF6 (0.01 mol/L) at 100 mV/s. (f) CV and DPV of 12+ (vs. Fc/Fc+).

Figure 2 (a) Solution-based ECD with 12+·2[OTf–] as an electrochromic material; Spectroelectrochemistry of 12+·2[OTf–], (b) first reduction, (c) second reduction; (d) UV–vis spectra of 12+, 1•+, 1, and 1•–. (e, f) Experimental (black line) and simulated (red line) EPR spectra of 12+ by 1, and 3 equiv. KC8, respectively. (g, h) The calculated spin density plots for 1•+ and 1•– (Isovalue 0.002).

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:

 下载:
下载: