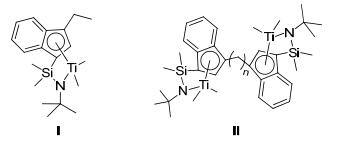
Figure 1.
Typical bridged (ansa) and bimetallic olefin polymerization catalysts
Synthesis of Titanium Heteroarylphosphinimine Complexes and Application for Ethylene Polymerization
Tieshi Wang , Jianjun Chen , Lin Ye , Aiying Zhang , Zengguo Feng

Nowadays, high-performance polyolefins paly an irreplaceable role in industrial and in our daily lives, [1] and have been extensive used in health and medical, food and specialty packaging, pipes and fittings, consumer and durable goods, and rigid packaging, among others.[2] In order to meet the growing demand of social development, numerous well-defined catalysts[3] with high activity and selectivity for polymerization based on group Ⅳ half- and nonmetallocene complexes have been designed, [4] which can control the polymerization performance in terms of branching type and amount, block structure, molecular weight and molecular weight distribution.[5] A dozen years ago, titanium or zirconium complexes using phosphinimine as ligands have been developed as a new family of olefin polymerization catalysts and performed with high activities, [6] which have already realized industrial application.[7]
As well know, the metal center has a decisive effect on the polymerization performances.[8] Especially, the bimetallic catalysts can exhibit marked metal-metal cooperative effect in contrast to their mono-metallic analogues[9] in how monomers are enchained to produce macromolecules, and so this effect apparently impacts the catalytic activity, product molecular weight and architecture, and comonomer enchaining fashion in the ethylene polymerization.[10]
Recently, Marks et al.[11] thoroughly described how bimetallic catalysts with identical group Ⅳ metal centers, and meta-metal proximity can dramatically modify the polyolefin molecular weight, chain branching, monomer repeat regioregularity. Different from their monometallic analogues (CGCTi1, Ⅰ, Figure 1), these bimetallic catalysts (such as, Cn-CGCTi2, Ⅱ, Figure 1) were bestowed with significant cooperative enchainment effects and unusual polymeric products possibly resulted from the proximate catalytic centers. However, studies toward the synthesis and activity of multinuclear phosphinimine complexes as olefin polymerization catalysts have been seldom carried out compared with other multinuclear half- and nonmetallocene complexes.[12]
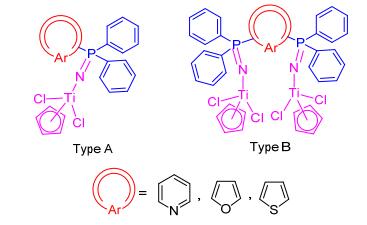
To this end, three kinds of bimetallic titanium catalysts containing heteroarylphosphinimine ligands (Type B, Figure 2) and their monometallic analogues (Type A, Figure 2) were designed and synthesised. These complexes exhibit different ligand architectures, electronic characteristics, and metal-metal proximities. A significant impact on their performance in the ethylene polymerization would be expected in this context.
To highlight the impact of steric and electronic differences in pyridine, thiophene and furan on the catalytic activity of Ti heteroarylphosphinimine complexes, two almost identical synthetic pathways were utilized to synthesize the mono- and bisdiphenylphosphine substituted heteroaryl compounds 1a~1c and 4a~4c according to the literature.[13] 2-Diphenylphosphanylpyridine (1a) and 2, 6-bis- (diphenylphosphanyl)pyridine (4a) were prepared from the substitution of 2-bromo- and 2, 6-dibromo-pyridine with diphenylphosphine using n-BuLi as a displacer, respectively. For the synthesis of other two monosubstituted heteroarylphosphines, thiophene and furan were first treated with an equivalent of n-BuLi and then in situ reacted with an equivalent of diphenylchlorophosphine to give rise to 2-(diphenylphosphanyl)thiophene (1b) and 2-(diphenyl- phosphanyl)furan (1c). However, the preparation of other two disubstituted heteroarylphosphines, thiophene and furan were first needed to treat not only with two equivalent amount of n-BuLi, but also with the same amount of N, N, N, N-tetramethylethylenediamine (TMEDA) as an auxiliary base followed by two equivalent amount of diphenylchlorophosphine to produce 2, 5-bis(diphenylphos- phanyl)thiophene (4b) and 2, 5-bis(diphenylphosphanyl)- furan (4c).
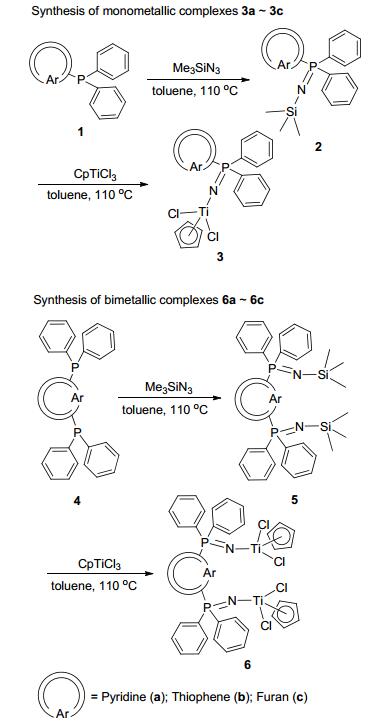
Subsequently, Staudinger reaction of the proceeding 1a~1c and 4a~4c with Me3SiN3 was conducted to form the corresponding heteroarylphosphinimine ligands 2a~2c and 5a~5c in excellent yields (92%~96%). Finally, these ligands were further reacted with CpTiCl3 in toluene to yield the mono- and bimetallic Ti half-metallocene catalysts 3a~3c and 6a~6c, respectively. Surprisingly, all the complexes could be purified by recrystallization in very high yields (85%~93%). Two synthetic routes (Routes A and B) of the Ti heteroarylphosphinimine halfmetallocene catalysts are schematized in Scheme 1.
The formulations of these species are all consistent with the observed 1H NMR and 13C NMR spectroscopic and elemental analytical data (See experimental section and supporting information).
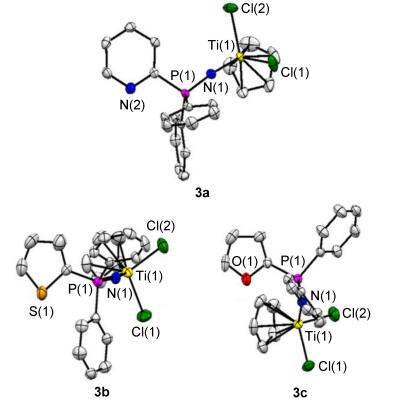
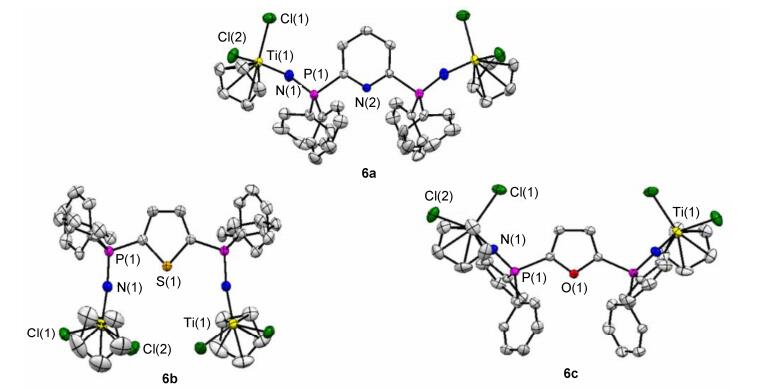
Single-crystal X-ray structure analysis was performed to determine the molecular structures of the resulting single and double Ti halfmetallocene catalysts 3a~3c and 6a~6c (CCDC No.: 1569213-1569218). Their ORTEP diagrams are shown in Figures 3 and 4, respectively, and selected data of bond lengths and bond angles are summarized in Table 1. In order to observe the structures conveniently, all the structural diagrams are given in three directions based on the direction of 2-pyridyl, 2-thienyl and 2-furyl and 2, 6-pyridyl, 2, 5-thienyl and 2, 5-furyl groups. All the structural data confirmed the formulation of both 3a~3c and 6a~6c and their metric parameters as expected.
 下载:
导出CSV
下载:
导出CSV
| 2-(Ph2PNTiCl2Cp)C5H4N (3a) | |||
| Ti(1)—N(1) | 1.779(3) | Cl(2)—Ti(1)—Cl(1) | 102.17(6) |
| P(1)—N(1) | 1.594(3) | P1—N(1)—Ti(1) | 167.10(18) |
| P(1)—C(6) | 1.809(3) | N(2)—C(6)—P(1) | 116.9(2) |
| P(1)—Cl(2) | 1.801(3) | N(1)—P(1)—C(6) | 111.70(15) |
| P(1)—C1(8) | 1.807(3) | N(1)—P(1)—C(12) | 113.31(15) |
| Ti(1)—Cl(1) | 2.3033(13) | N(1)—P(1)—C(18) | 109.84(15) |
| Ti(1)—Cl(2) | 2.2955(13) | N(1)—Ti(1)—Cpa | 120.329(106) |
| 2, 6-(Ph2PNTiCl2Cp)2C5H3N (6a) | |||
| Ti(1)—N(1) | 1.784(3) | Cl(2)—Ti(1)—Cl(1) | 101.52(4) |
| P(2)—N(1) | 1.578(3) | P(2)—N(1)—Ti(1) | 161.75(19) |
| P(2)—C6 | 1.800(3) | N(2)—C(12)—P(2) | 114.7(2) |
| P(2)—C12 | 1.821(3) | N(1)—P(2)—C(6) | 114.76(15) |
| P(2)—C15 | 1.800(3) | N(1)—P(2)—C(12) | 108.69(15) |
| Ti(1)—Cl(1) | 2.3014(11) | N(1)—P(2)—C(15) | 111.07(15) |
| Ti(1)—Cl(2) | 2.2900(11) | N1—Ti(1)—Cpa | 117.769(93) |
| 2-(Ph2PNTiCl2Cp)2C4H3S (3b) | |||
| Ti(1)—N(1) | 1.775(4) | Cl(2)—Ti(1)—Cl(1) | 102.55(6) |
| P1—N(1) | 1.590(4) | P1—N(1)—Ti(1) | 164.3(2) |
| P(1)—C4 | 1.781(4) | S(1)—C(4)—P(1) | 122.0(2) |
| P(1)—C5 | 1.800(4) | N(1)—P(1)—C(4) | 114.45(19) |
| P(1)—C11 | 1.795(4) | N(1)—P(1)—C(5) | 111.60(19) |
| Ti(1)—Cl(1) | 2.3184(14) | N(1)—P(1)—C(11) | 110.78(19) |
| Ti(1)—Cl(2) | 2.2963(15) | N1—Ti(1)—Cpa | 120.213(132) |
| 2, 5-(Ph2PNTiCl2Cp)2C4H2S (6b) | |||
| Ti(1)—N(1) | 1.771(4) | Cl(2)—Ti(1)—Cl(1) | 101.65(7) |
| P1—N(1) | 1.596(4) | P(1)—N(1)—Ti(1) | 162.2(2) |
| P(1)—C1 | 1.800(4) | S(1)—C(13)—P(1) | 118.8(2) |
| P(1)—C7 | 1.789(4) | N(1)—P(1)—C(1) | 113.97(19) |
| P(1)—C13 | 1.792(4) | N(1)—P(1)—C(7) | 109.92(19) |
| Ti(1)—Cl(1) | 2.3033(14) | N(1)—P(1)—C(13) | 109.14(19) |
| Ti(1)—Cl(2) | 2.2906(17) | N1—Ti(1)—Cpa | 118.233(119) |
| 2-(Ph2PNTiCl2Cp)C4H3O (3c) | |||
| Ti(1)—N(1) | 1.778(2) | Cl(1)—Ti(1)—Cl(2) | 101.98(4) |
| P1—N(1) | 1.587(2) | P(1)—N(1)—Ti(1) | 162.99(15) |
| P(1)—C(1) | 1.782(3) | O(1)—C(1)—P(1) | 116.09(19) |
| P(1)—C(5) | 1.791(3) | N(1)—P(1)—C(1) | 114.22(12) |
| P(1)—C(11) | 1.801(3) | N(1)—P(1)—C(5) | 111.70(12) |
| Ti(1)—Cl(1) | 2.3021(10) | N(1)—P(1)—C1(1) | 111.84(12) |
| Ti(1)—Cl(2) | 2.3093(9) | N1—Ti(1)—Cpa | 119.630(73) |
| 2, 5-(Ph2PNTiCl2Cp)2C4H2O (6c) | |||
| Ti(1)—N(1) | 1.791(2) | Cl(2)—Ti(1)—Cl(1) | 101.40(4) |
| P1—N(1) | 1.576(2) | P(1)—N(1)—Ti(1) | 161.91(15) |
| P(1)—C(1) | 1.793(3) | O(1)—C(13)—P(1) | 118.47(17) |
| P(1)—C(7) | 1.801(3) | N(1)—P(1)—C(1) | 117.63(12) |
| P(1)—C(13) | 1.792(3) | N(1)—P(1)—C(7) | 110.74(12) |
| Ti(1)—Cl(1) | 2.3042(9) | N(1)—P(1)—C(13) | 107.73(12) |
| Ti(1)—Cl(2) | 2.2922(10) | N1—Ti(1)—Cpa | 118.739(76) |
| a N—Ti—Cp angles refer to the angle at Ti between N and the centroid of the five η5-carbons of the Cp-type ligands. | |||
At the same time, in each complex, the geometry around the N atom is proximately analogue, and the P—N and Ti—N distances are very similar, averaging 1.587(3) and 1.780(3) Å, respectively, which closely corresponded to the values of 1.560(1) and 1.780(1) Å found in the parent complex CpCl2TiNPPh3.[14] However, the P—N—Ti angles ranging between 161.75(19)° and 167.10 (18)° are slightly smaller than the reported 174.7(9)°.[14] Herein the similar bond distances and slightly deviated bond angles are highly consistent with the steric demands and potential energy influence of the substituted heteroarylphosphinimine lig-ands. Meanwhile, compared to the previous report, [14] the Cl—Ti—Cl and N—Ti—Cl angles are slightly Hydrogen atoms have been omitted for clarity different, averaging 101.9(5)° and 102.9(10)°, respectively.
The steric demands of all the heteroarylphosphinimine ligands can be embodied by the angles at Ti between the centroid of the η5-carbons of the Cp-type ligands and N atom.[15] For the asymmetric single Ti heteroarylphos- phinimine complexes 3a~3c, the N—Ti—Cp angles range from 119.6 (73)° to 120.3 (106)° similar to that seen in CpCl2TiNPPh3 120.9 (7)°.[12, e] These values are slightly decreased to a range of 117.8 (93)° and 118.7 (76)° for the symmetric double Ti halfmetallocene catalysts 6a~6c, indicating a predominantly steric influence of heteroar- ylphosphinimine and cyclopentadienyl ligands.
Moreover, an analysis on the crystal structures of 6a~6c as shown in Figure 3 and a comparison to referenced compounds reported in the literature, [13c] clearly suggested these double Ti heteroarylphosphinimine complexes possessing a quite close C2 symmetry in solid states with the fold axis passing through the heteroatoms (N, S, O) and bisecting their endocyclic angle. For the single Ti heteroarylphosphinimine complexes 3a~3c, a slight distortion from N atom geometry was observed, probably due to the tilt to avoid steric interaction. Moreover, the CpTiCl2 groups are oppositely positioned in the both wings of heteroarylcyclics in 6a and 6c. However, these groups locate in the same side of heteroarylcyclics in 6b. Consequently, the Ti—Ti distance is only 6.732(21) Å for 6b, meaning that both metal centres may reach a close cooperative interaction.[13b] Conversely, in 6a and 6c this intramolecular cooperativity is not possible because of the large Ti—Ti distance of 11.076(25) and 10.540(25) Å, respectively. The different spatial orientations of the CpTiCl2 groups and different Ti—Ti distances in 6a~6c revealed different steric demands and electronic effects from pyridine to thiophene and furan. Consequently, it would lend further credence to the potential applications of symmetric bimetallic Ti halfmetallocene catalyst 6a~6c.
The catalytic activities of both mono- and bimetallic titanium heteroarylphosphinimine complexes 3a~3c and 6a~6c toward the ethylene polymerizations were demonstrated using methylaluminoxane (MAO) as cocatalyst at a ratio of Al/Ti=600 and under 0.5 MPa of ethylene. The detailed polymerization conditions and results are summarized in Table 2. In all the cases, temperature automatically rose by 5~10 ℃ due to heat release of ethylene poly- merization. As can be seen, mononuclear catalysts 3a~3c displayed high catalytic activity, and the resulting polyethylene had relatively high molecular weight, but showed broad molecular weight distributions. One reasonable explanation for this phenomenon is that, the very high molecular weight polyethylene produced in initial period of polymerization due to poor solubility and quick precipitation hindered the contact of ethylene monomer with the active Ti centres, resulting in the inhomogeneity at the later stage of polymerization, which certainly leads to the formation of broad molecular weight distribution. The DSC data showed that the polyethylene samples have high melting points (> 140 ℃), indicating that the polymers obtained are linear polyethylene with high crystallinity.[17]
下载:
导出CSV
| Entry | Cat. | Cat./μmol | T/℃ | Time/min | Yield/g | Activityb×106 | Tmc/℃ | Mnd×104 | Mwd×104 | PDId |
| 1 | 3a | 20 | 50 | 15 | 2.15 | 0.43 | 142.7 | 9.44 | 68.10 | 7.2 |
| 2 | 3b | 20 | 50 | 15 | 4.25 | 0.85 | 141.5 | 7.04 | 58.19 | 8.3 |
| 3 | 3c | 20 | 50 | 15 | 3.16 | 0.63 | 140.7 | 5.86 | 52.26 | 8.9 |
| 4 | 6a | 10 | 50 | 15 | 3.21 | 1.28 | 140.5 | 5.60 | 47.42 | 8.5 |
| 5 | 6b | 10 | 50 | 15 | 4.69 | 1.88 | 140.5 | 10.26 | 48.31 | 4.7 |
| 6 | 6c | 10 | 50 | 15 | 3.15 | 1.26 | 143.1 | 5.59 | 66.97 | 12.0 |
| 7 | 6b | 10 | 40 | 15 | 3.45 | 1.38 | 143.6 | 8.52 | 121.7 | 14.3 |
| 8 | 6b | 10 | 30 | 15 | 3.31 | 1.32 | 145.3 | 10.75 | 179.7 | 16.7 |
| 9 | 6b | 10 | 30 | 5 | 2.42 | 2.90 | 144.1 | 9.59 | 172.3 | 18.0 |
| a Polymerization conditions: 150 mL stainless steel autoclave, 100 mL of toluene, under 0.5 MPa. b Activity in units of g (mol•cat.)-1 h-1. c Melting temperature was determined by differential scanning of calorimetry (DSC). d Determined by GPC at 160 ℃ in trichlorobenzene using polystyrene standards, given in units of g• mol-1. | ||||||||||
The activity of bimetallic complexes [6a~6c, 1.26~1.88×106 g (mol•cat.)-1•h-1] is significantly higher than that of monometallic ones [3a~3c, 0.43~0.85×106 g (mol•cat.)-1•h-1] under identical polymerization conditions, presumably due to the greater steric hindrance exerted by the shorter bridge lead to the termination kinetics substantially depressed with the significant Mw enhancement.[11] However, the polyethylene samples obtained by 6a~6c exhibited a wider molecular weight distribution, consistent with the catalytic properties of bimetallic compounds reported in literature.[18] The catalytic activity of thiophene containing catalysts is higher than that of pyridine and furan derived catalysts, the same phenomenon was observed for both mononuclear and binuclear catalysts, indicating that the thiophene-base catalysts has more advantages. Therefore, a series of polymerization experi- ments were carried out at different temperatures and reaction times to gain insight into the polymerization properties of 6b. It was found that 6b can produce ultrahigh Mw polyethylene at a lower polymerization temperature (179.7×104 g•mol-1) compared to at a higher temperature (48.31×104 g•mol-1), together with a significantly bimodal molecular weight distributions. Furthermore, the molecular weight and molecular weight distribution of polyethylenes obtained change a little under different reaction time (Entry 8 vs Entry 9), but the longer polymerization time can increase the polymer yield. These results clearly demonstrated that 6b has a good thermal stability and life-time, and polymerization products with different structural properties could be obtained by controlling the polymerization conditions, especially bimodal ultrahigh Mw polyethylene with a broad application prospect, due to their good machinability and excellent mechanical properties.[16] A more detailed investigation on the effects of these monometallic and bimetallic titanium heteroaryl-phos- phinimine complexes on the ethylene copolymerization with other encumbered olefins is underway.
Both mono- and bimetallic Ti heteroarylphosphinimine R-PPh2(NTiCpCl2) (R=2-pyridyl, 2-thienyl and 2-furyl) and (Cl2CpTiN)-Ph2P-R′-PPh2(NTiCpCl2) (R'=2, 6-pyri- dyl, 2, 5-thienyl and 2, 5-furyl) were synthesized and characterized by means of 1H NMR and 13C NMR spectroscopic methods and further confirmed by single-crystal X-ray diffraction analysis. These bimetallic heterocycle containing complexes have a C2 symmetrical structure in solid states, where a Ti—Ti distance of 6.732(21) Å in 6b is substantially shorter than that of 11.076(25) and 10.540(25) Å in 6a and 6c, meaning that two metal atoms may reach a close cooperative interaction in the polymerization process. After be activated by MAO, these bimetallic Ti phosphinimine complexes displayed a higher catalytic activity to ethylene polymerization compared with the monometallic analogues. Especially, 6b can produce ultrahigh Mw polyethylene have a bimodal molecular weight distributions at lower polymerization temperature.
All synthetic experiments were carried out in a nitrogen atmosphere glovebox (MBRAUN LABstar 5470, O2 < 0.1 mg/L, H2O < 0.1 mg/L). All chemicals purchased were purified by standard purification procedures. Tetrahydrofuran (THF), pentane and toluene were distilled over sodium and benzophenone under nitrogen atmosphere and were stored in a Schlenk tube in the glovebox. Ethylene (purity > 99.99%) and methylalumoxane (MAO) were provided by Sinopec and were used as received. All other chemicals were purchased from Tokyo Chemical Industry (Shanghai).
All 1H NMR, 13C NMR, and 31P NMR spectra were recorded on a Bruker Ascend 400 M spectrometer and were recorded in C6D6 at 25 ℃ unless otherwise noted. 1H NMR and 13C NMR chemical shifts are given in δ and are referenced to tetramethylsilane. 31P spectra are referenced to external 85% H3PO4, and chemical shifts are reported in units of δ. Elemental analyses were performed by the element analysis system (Eurovector EA3000). Molecular weights and molecular weight distributions of the resultant polymers were measured by the gel permeation chromatography (GPC, Waters 2000, America) using trichlorobenzene as solvent at 160 ℃. The molecular weight was calculated by a standard procedure based on the calibration with standard polystyrene samples. Thermal properties of obtained olefin polymers were investigated on a differential scanning calorimeter (DSC-60, Shimazdu), and under nitrogen atmosphere with the heating rate of 10 ℃/min. X-ray diffraction data were collected by a Bruker APEX Ⅱ DUO diffractometer (graphite-monochromatized Mo Kα radiation, λ=0.71073 Å) at 153 K. The hemisphere of reflection data was collected using ω scan mode. The structures were solved by direct methods and refined by full matrix least squares techniques with anisotropic parameters by SHELXTL program. All hydrogen atoms were placed in calculated positions and refined riding on the corresponding carbon atoms with isotropic thermal parameters.
2-Bromopyridine (1.58 g, 0.01 mol) was diluted in hexane (100 mL) and cooled to -78 ℃ in a cold bath of liquid nitrogen/acetone. n-BuLi (6.25 mL of 1.6 mol•L-1 solution in hexane) was added dropwise causing an colour change from colourless to dark red. The solution was stirred at -78 ℃ for 1 h. Diphenylphosphine (2.21 g, 0.01 mol) was added and then the solution was warmed to room temperature slowly. After the mixture was stirred overnight, water (10 mL) was added and the colour changed from dark red to pale yellow. The organic phase was dried over MgSO4. The solution was filtered and the solvent was removed in vacuo. The yellowish oil was purified by column chromatography on silica gel [eluent: V(hexane):V(AcOEt)=9:1]. The solvents were removed in vacuo and a white powder was obtained 1a. 2.18 g, yield 83%. m.p. 82~83 ℃; 1H NMR (400 MHz, C6D6) δ: 8.50 (d, J=4.6 Hz, 1H, py), 7.52 (t, J=7.2 Hz, 4H, o-PPh2), 7.10~6.94 (m, 7H, m-PPh2, p-PPh2, py), 6.85 (t, J=7.6 Hz, 1H, py), 6.48 (t, J=6.0 Hz, 1H, py); 13C NMR (101 MHz, C6D6) δ: 164.3 (d, J=1.9 Hz), 150.21 (d, J=11.8 Hz), 137.14 (d, J=11.1 Hz), 134.87 (d, J=3.4 Hz), 134.48 (s), 134.28 (s), 128.7 (s), 128.39 (d, J=7.1 Hz), 121.61 (s); 31P NMR (162 MHz, C6D6) δ: -1.99 (s). Anal. calcd for C17H14NP: C 77.56, H 5.36, N 5.32; found C 77.24, H 5.36, N 5.29.
To a stirred THF solution (100 mL) of diphenylphosphine (3.73 g, 0.02 mol) was added n-BuLi (12.5 mL, 1.6 mol•L-1 in hexanes) at -78 ℃ and the deep red solution was stirred overnight at room temperature. To this solution was added dropwise a 20 mL THF solution of 2, 6-dibromopyridine (2.36 g, 0.01 mol). After the mixture was stirred overnight, the solvent was removed in vacuo and water (30 mL) was added to quench the reaction. Then the mixture was extracted three times with CH2Cl2. The organic phases were combined, washed with saturated NaHCO3 solution, and dried with MgSO4. The solution was filtered and the solvent was removed in vacuo, yielded a colourless oily solid 4a, which was recrystallized with CH2Cl2 and hexane. 3.89 g, yield 78%. m.p. 133~134 ℃; 1H NMR (400 MHz, C6D6) δ: 7.44 (m, 8H, o-PPh2), 7.02 (m, 12H, m-PPh2, p-PPh2), 6.94 (d, J=8.4 Hz, 2H, py), 6.73 (m, 1H, py); 13C NMR (101 MHz, C6D6) δ: 164.79 (d, J=7 Hz), 137.04 (d, J=12 Hz), 134.75 (t, J=9.3 Hz), 134.39 (t, J=20.3 Hz), 128.59 (s), 128.32 (t, J=7.5 Hz), 126.71 (d, J=25.5 Hz); 31P NMR (162 MHz, C6D6) δ: -1.96 (s). Anal. calcd for C29H23NP2: C 77.84, H 5.18, N 3.13; found C 77.81, H 5.15, N 3.18.
A solution of thiophene (1.68 g, 0.02 mol) in 100 mL of dry Et2O was cooled to -78 ℃, and n-BuLi (12.5 mL, 1.6 mol•L-1 in hexanes) was added dropwise using a syringe. The solution was stirred at room temperature for 2 h, resulted in a white suspension mixture, and then cooled again to -78 ℃. Diphenylchlorophosphine (4.41 g, 0.02 mol) was added dropwise to the suspension mixture. The mixture was gradually warmed to room temperature and stirred overnight. The mixture was then poured into water (100 mL) and extracted using CH2Cl2 (100 mL×3). The organic phases combined, washed with NaCl solution and dried with MgSO4. Removal of the solvent yielded a viscous oil, which was purified by column chromatography using silica gel and [V(CH2Cl2):V(hexanes)=1:4] as eluent. The product was then recrystallized in CH2Cl2- methanol and obtained as a colorless crystal. 4.11 g, yield 76%. m.p. 43~44 ℃; 1H NMR (400 MHz, C6D6) δ: 7.43~7.38 (m, 4H, o-PPh2), 7.22~7.17 (m, 1H, tp), 7.04~6.95 (m, 7H, m-PPh2, p-PPh2, tp), 6.73~6.68 (m, 1H, tp); 13C NMR (101 MHz, C6D6) δ: 138.46~138.17 (dd, J=8.1, 18.2 Hz), 136.42 (s), 136.16 (s), 133.13 (d, J=19.9 Hz), 131.83 (s), 128.61 (s), 128.41 (d, J=7.1 Hz); 31P NMR (162 MHz, C6D6) δ: -20.10 (s). Anal. calcd for C16H13PS: C 71.62, H 4.88; found C 71.46, H 4.91.
A solution of thiophene (1.68 g, 0.02 mol) and tetramethylethylenediamine (TMEDA) (4.75 g, 0.04 mol) in 100 mL dry hexanes was cooled to 0 ℃, and n-BuLi (25 mL, 1.6 mol•L-1 in hexanes) was added dropwise using a syringe. Then the reaction mixture was refluxed for 3 h and the resultant white suspension was cooled to 0 ℃ again. Diphenylchlorophosphine (8.83 g, 0.04 mol) was added slowly and the reaction mixture was allowed to warm to room temperature with stirring overnight. Water (50 mL) was added to the mixture, which was then extracted three times with CH2Cl2 (100 mL×3). The organic phases were combined, washed with NaCl solution, and dried with MgSO4. The solvent was then removed in vacuo, yielding a crude product, which was purified by column chromatography on silica gel with [V(CH2Cl2):V(hexanes)=1:3] as eluent. The residue was dissolved in the minimum amount of hexane and stored at -30 ℃ to precipitate colourless crystals. 7.06 g, yield 78%. m.p. 68~69 ℃; 1H NMR (400 MHz, C6D6) δ: 7.438~7.375 (m, 8H, o-PPh2), 7.16 (m, 2H, tp), 7.01~6.96 (m, 12H, m-PPh2, p-PPh2); 13C NMR (101 MHz, C6D6) δ: 146.00 (d, J=31.3 Hz), 138.02 (d, J=10.0 Hz), 137.08 (dd, J=23.0, 6.6 Hz), 133.56 (d, J=20.2 Hz), 128.97 (s), 128.66 (d, J=7.1 Hz); 31P NMR (162 MHz, C6D6) δ: -18.77 (s). Anal. calcd for C28H22P2S: C 74.32, H 4.90; found C 74.21, H 4.96.
A solution of n-BuLi (12.5 mL, 1.6 mol•L-1 in hexane) was slowly added to furan (1.36 g, 0.02 mol, in 100 mL THF) at -78 ℃. The reaction mixture was allowed to warm to room temperature and stirring was continued for 20 min. Diphenylchlorophosphine (4.41 g, 0.02 mol) was slowly added at -78 ℃ and the reaction mixture was allowed to warm to room temperature and stirred for 12 h. Water (50 mL) was added to the mixture, which was then extracted three times with CH2Cl2 (100 mL×3). The organic phases combined, washed with NaCl solution, and dried with MgSO4. The solvent was then removed in vacuo, yielding a crude product, which was purified by column chromatography on silica gel with [V(CH2Cl2):V(hexanes)=1:3] as eluent. The solvents were removed in vacuo and a white powder was obtained. 4.34 g, yield 86%. m.p. 38~39 ℃; 1H NMR (400 MHz, C6D6) δ: 7.48 (m, 4H, o-PPh2), 7.18 (s, 1H, fn), 7.02 (m, 6H, m-PPh2, p-PPh2), 6.58 (s, 1H, fn), 6.00 (s, 1H, fn); 13C NMR (101 MHz, C6D6) δ: 152.74 (d, J=19.1 Hz), 147.39 (d, J=1.5 Hz), 136.73 (d, J=6.6 Hz), 133.42 (d, J=19.7 Hz), 128.62 (s), 128.39 (d, J=7.0 Hz), 121.86 (d, J=24.5 Hz), 110.52 (d, J=5.9 Hz); 31P NMR (162 MHz, C6D6) δ: -26.96 (s). Anal. calcd for C16H13OP: C 76.18, H 5.19; found C 75.95, H 5.21.
The preparation of 2, 5-bis(diphenylphosphanyl)furan was based on a modified literature procedure.[12, c] A solution of furan (1.36 g, 0.02 mol) in dry diethyl ether (80 mL) was cooled to -78 ℃, and t-BuLi (26.7 mL, 1.5 mol•L-1 in pentane) was added dropwise, using a syringe. The reaction mixture was stirred for 1 h at -78 ℃ and then allowed to warm to room temperature slowly. The mixture was stirred for additional 1.5 h at room temperature and got a white suspension. Diphenylchlorophosphine (8.83 g, 0.04 mol) was slowly added at -78 ℃ and the reaction mixture was allowed to warm to room temperature and stirred for overnight. The solvent was removed in vacuo and water (30 mL) was added to quench the reaction. The mixture was extracted three times with CH2Cl2. The organic phases were combined, washed with saturated NaHCO3 solution, and dried with MgSO4. The solution was filtered and the solvent was removed in vacuo, yielded a colourless oily solid, which was recrystallized with CH2Cl2 and hexane. 7.16 g, yield 82%. m.p. 154~155 ℃; 1H NMR (400 MHz, C6D6) δ: 7.43~7.38 (m, 8H, o-PPh2), 7.03~6.96 (m, 12H, m-PPh2, p-PPh2), 6.58 (t, 2H, fn); 13C NMR (101 MHz, C6D6) δ: 158.64 (d, J=25.3 Hz), 136.66 (d, J=6.7 Hz), 133.71 (d, J=19.8 Hz), 128.77 (s), 128.58 (d, J=7.1 Hz), 122.93 (dd, J=28.1, 6.7 Hz); 31P NMR (162 MHz, C6D6) δ: -26.9 (s). Anal. calcd for C28H22- OP2: C 77.06, H 5.08; found C 77.12, H 5.07.
The Compounds 2a~2c and 5a~5c were prepared using a modification of the literature procedure.[5d] Only one preparation is described here.
N3SiMe3 (1.73 g, 0.015 mol) was added to a solution of 2-(diphenylphosphanyl)pyridine (1a) (2.63 g, 0.01 mol) in 20 mL of toluene slowly, and the reaction mixture was heated at reflux for 12 h. When the solvent and excess of N3SiMe3 was removed under vacuum 2-(Ph2P=NTMS) pyridine (2a), was obtained. 3.36 g, yield 96%, white solid. m.p. 90~91 ℃; 1H NMR (400 MHz, C6D6) δ: 8.38 (m, 1H, py), 8.28 (m, 1H, py), 7.98~7.87 (m, 4H, o-PPh2), 7.04~6.95 (m, 8H, m-PPh2, p-PPh2, py), 6.45~6.41 (m, 1H, py), 0.34 (s, 9H, SiMe3); 13C NMR (101 MHz, C6D6) δ: 159.96 (s), 158.61 (s), 149.52 (d, J=20.2 Hz), 136.19 (s), 135.47 (d, J=9 Hz), 134.69 (d, J=20 Hz), 132.6 (d, J=10.1 Hz), 130.79 (d, J=3 Hz), 124.01 (d, J=3 Hz), 4.31 (d, J=2.8 Hz); 31P NMR (162 MHz, C6D6) δ: -4.97 (s). Anal. calcd for C20H23N2PSi: C 68.54, H 6.61, N 7.99; found C 68.39, H 6.60, N 7.78.
2, 6-Bis(Ph2P=NTMS)2pyridine (5a): 2.96 g, yield 95%. White solid, m.p. 126~127 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.45~8.37 (m, 2H, py), 8.05~7.97 (m, 1H, py), 7.51~7.43 (m, 12H, m-PPh2, p-PPh2), 7.27~7.22 (m, 8H, o-PPh2), 0.01 (s, 18H, SiMe3); 13C NMR (101 MHz, C6D6) δ: 159.74 (d, J=17.7 Hz), 158.42 (d, J=17.6 Hz), 135.71 (t, J=8.6 Hz), 135.38 (s), 134.32 (d, J=11.8 Hz), 132.27~131.97 (m), 130.39 (s), 4.04 (d, J=2.7 Hz); 31P NMR (162 MHz, C6D6) δ: -5.38 (s). Anal. calcd for C35H41N3P2Si2: C 67.60, H 6.65, N 6.76; found C 67.43, H 6.72, N 6.65.
2-(Ph2P=NTMS)thiophene (2b): 1.64 g, yield 92%. White solid, m.p. 95~96 ℃; 1H NMR (400 MHz, C6D6) δ: 7.70 (m, 4H, o-PPh2), 7.17 (m, 1H, tp), 6.98 (s, 7H, m-PPh2, p-PPh2, tp), 6.60 (s, 1H, tp), 0.33 (s, 9H, SiMe3); 13C NMR (101 MHz, C6D6) δ: 139.07 (s), 138.01 (s), 136.81 (s), 135.73 (s), 135.23 (d, J=10.0 Hz), 132.26 (d, J=4.6 Hz), 131.61 (d, J=10.8 Hz), 130.75 (d, J=2.9 Hz), 3.95 (d, J=3.4 Hz); 31P NMR (162 MHz, C6D6) δ: -9.80 (s). Anal. calcd for C19H22NPSSi: C 64.19, H 6.24, N 3.94; found C 63.98, H 6.11, N 3.88.
2, 5-Bis(Ph2P=NTMS)2thiophene (5b): 2.98 g, yield 95%. White solid, m.p. 99~100 ℃; 1H NMR (400 MHz, C6D6) δ: 7.56~7.44 (m, 8H, o-PPh2), 7.01~6.96 (m, 2H, tp), 6.85~6.72 (m, 12H, m-PPh2, p-PPh2), 0.15 (s, 18H, SiMe3); 13C NMR (101 MHz, C6D6) δ: 146.16 (dd, J=99.2, 2.6 Hz), 136.13 (s), 135.64~135.32 (m), 135.05 (s), 131.55 (d, J=11.2 Hz), 130.97 (s), 128.14 (s), 3.87 (d, J=3.2 Hz); 31P NMR (162 MHz, C6D6) δ: -10.44 (s). Anal. calcd for C34H40N2P2SSi2: C 65.14, H 6.43, N 4.47; found C 64.85, H 6.29, N 4.51.
2-(Ph2P=NTMS)furan (2c): 1.63 g, yield 96%. White solid, m.p. 88~89 ℃; 1H NMR (400 MHz, C6D6) δ: 7.81 (m, 4H, o-PPh2), 7.05 (m, 6H, fn, m-PPh2, p-PPh2), 6.84 (s, 1H, fn), 5.95 (s, 1H, fn), 0.39 (s, 9H, SiMe3); 13C NMR (101 MHz, C6D6) δ: 151.54 (d, J=127.2 Hz), 146.95 (d, J=6.8 Hz), 135.63 (s), 134.53 (s), 131.54 (d, J=10.9 Hz), 130.84 (d, J=2.8 Hz), 121.58 (d, J=18.1 Hz), 110.30 (d, J=8.1 Hz), 3.84 (d, J=3.6 Hz); 31P NMR (162 MHz, C6D6) δ: -14.08 (s). Anal. calcd for C19H22NOPSi: C 67.23, H 6.53, N 4.13; found C 67.41, H 6.45, N 4.21.
2, 5-Bis(Ph2P=NTMS)2furan (5c): 2.87 g, yield 94%. White solid, m.p. 67~68 ℃; 1H NMR (400 MHz, C6D6) δ: 7.58~7.52 (m, 8H, o-PPh2), 6.89 (m, 12H, m-PPh2, p-PPh2), 6.73 (s, 2H, fn), 0.21 (s, 18H, SiMe3); 13C NMR (101 MHz, C6D6) δ: 156.80 (dd, J=121.0, 4.3 Hz), 135.07 (d, J=112.1 Hz), 133.38 (d, J=19.9 Hz), 131.45 (d, J=11.0 Hz), 130.91 (d, J=2.9 Hz), 121.78 (dd, J=17.8, 8.1 Hz), 3.88 (d, J=3.7 Hz); 31P NMR (162 MHz, C6D6) δ: -14.52 (s). Anal. calcd for C34H40N2OP2Si2: C 66.86, H 6.60, N 4.59; found C 66.78, H 6.73, N 4.55.
Compounds 3a~3c and 6a~6c were prepared by a modification of a literature procedure.[5, d] Only one preparation is described here.
To a solution of CpTiCl3 (0.22 g, 1 mmol) in 15 mL toluene was added 2-(Ph2P=NTMS)pyridine (0.35 g, 1 mmol) slowly at room temperature. Then the mixture solution was refluxed for 12 h at 110 ℃. The solvent was removed under vacuum, and the crude product was purified by washing with hexane (20 mL×3) and drying under vacuum, 2-(Ph2PNTiCl2Cp)C5H4N (3a) was collected. 0.43 g, yield 93%. Yellow powder, m.p. 203~204 ℃; 1H NMR (400 MHz, C6D6) δ: 8.96 (m, 1H, py), 8.13 (d, J=4.6 Hz, 1H, py), 7.95~7.83 (m, 4H, o-PPh2), 7.07~6.93 (m, 6H, m-PPh2, p-PPh2), 6.46~6.38 (m, 2H, py), 6.20 (s, 5H, Cp); 13C NMR (101 MHz, C6D6) δ: 150.01 (d, J=20.5 Hz), 136.60 (d, J=10.0 Hz), 132.93 (d, J=10.2 Hz), 132.49 (d, J=2.9 Hz), 130.96 (s), 130.74 (s), 130.45 (s), 129.46 (s), 128.78 (d, J=12.6 Hz), 125.56 (d, J=3.6 Hz), 119.15 (s), 115.37 (s); 31P NMR (162 MHz, C6D6) δ: -3.02 (s). Anal. calcd for C22H19Cl2N2PTi: C 57.30, H 4.15, N 6.07; found C 57.39, H 4.12, N 6.13.
2, 6-(Ph2PNTiCl2Cp)2C5H3N (6a): 0.73 g, yield 87%. Yellow powder, m.p. 254~255 ℃; 1H NMR (400 MHz, CD2Cl2) δ: 8.81 (m, 2H, py), 8.27 (m, 1H, py), 7.75~7.57 (m, 12H, m-PPh2, p-PPh2), 7.49~7.39 (m, 8H, o-PPh2), 6.28 (s, 10H, Cp); 13C NMR (101 MHz, CD2Cl2) δ: 155.78 (d, J=18.6 Hz), 138.16~137.92 (m), 133.09 (dd, J=15.6, 6.9 Hz), 132.52 (d, J=10.5 Hz), 128.90 (dd, J=15.8, 11.1 Hz), 127.94 (d, J=38.0 Hz), 125.20 (s), 115.82 (s); 31P NMR (162 MHz, CD2Cl2) δ: -2.07 (s). Anal. calcd for C39H33Cl4N3P2Ti2: C 55.55, H 3.94, N 4.98; found C 55.71, H 4.06, N 5.04.
2-(Ph2PNTiCl2Cp)C4H3S (3b): 0.43 g, yield 92%. Yellow powder, m.p. 203~204 ℃; 1H NMR (400 MHz, C6D6) δ: 7.66 (m, 4H, o-PPh2), 7.36 (m, 1H, tp), 6.92~6.79 (m, 7H, m-PPh2, p-PPh2, tp), 6.45 (m, 1H, tp), 6.15 (s, 5H, Cp); 13C NMR (101 MHz, C6D6) δ: 138.56 (d, J=10.3 Hz), 134.71 (d, J=5.0 Hz), 132.43 (d, J=2.9 Hz), 132.12 (d, J=11.1 Hz), 130.96 (s), 129.91 (s), 128.69 (d, J=13.1 Hz), 128.50 (s), 118.94 (s), 115.24 (s); 31P NMR (162 MHz, C6D6) δ: -6.66 (s). Anal. calcd for C21H18Cl2- NPSTi: C 54.11, H 3.89, N 3.00; found C 53.96, H 4.01, N 2.91.
2, 5-(Ph2PNTiCl2Cp)2C4H2S (6b): 0.72 g, yield 85%. m.p. 241~242 ℃; 1H NMR (400 MHz, CD2Cl2) δ: 7.81~7.64 (m, 10H, o-PPh2, tp), 7.64~7.55 (m, 4H, p-PPh2), 7.51 (m, 8H, m-PPh2), 6.21 (s, 5H, Cp); 13C NMR (101 MHz, CD2Cl2) δ: 140.72 (dd, J=103.2, 3.5 Hz), 139.02~138.15 (m), 133.54 (s), 132.16 (d, J=11.5 Hz), 129.32 (d, J=13.5 Hz), 128.39 (s), 119.17 (s), 115.95 (s); 31P NMR (162 MHz, C6D6) δ: -5.12 (s). Anal. calcd for C38H32Cl4- N2P2STi2: C 53.81, H 3.80, N 3.30; found C 53.94, H 3.93, N 3.34.
2-(Ph2PNTiCl2Cp)C4H3O (3c): 0.40 g, yield 89%. Yellow powder, m.p. 206~207 ℃; 1H NMR (400 MHz, C6D6) δ: 7.66~7.61 (m, 4H, o-PPh2), 7.22 (s, 1H, fn), 6.93~6.79 (m, 7H, m-PPh2, p-PPh2, fn), 6.14 (s, 5H, Cp), 5.78~5.66 (m, 1H, fn); 13C NMR (101 MHz, C6D6) δ: 148.73 (d, J=7.6 Hz), 132.55 (d, J=3.0 Hz), 132.01 (d, J=11.2 Hz), 128.76 (d, J=13.2 Hz), 128.61 (s), 125.76 (d, J=19.0 Hz), 118.94 (s), 115.30 (s), 111.24 (d, J=9.0 Hz); 31P NMR (162 MHz, C6D6) δ: -11.9 (s). Anal. calcd for C21H18Cl2NOPTi: C 56.04, H 4.03, N 3.11; found C 56.13, H 4.11, N 3.13.
2, 5-(Ph2PNTiCl2Cp)2C4H2O (6c): 0.76 g, yield 91%. Yellow powder, m.p. 224~225 ℃; 1H NMR (400 MHz, C6D6) δ: 7.66~7.57 (m, 8H, o-PPh2), 7.00~6.92 (m, 14H, m-PPh2, p-PPh2, fn), 6.18 (s, 10H, Cp); 13C NMR (101 MHz, C6D6) δ: 137.52 (s), 132.87 (s), 131.98 (d, J=11.4 Hz), 129.10~128.84 (m), 128.20 (s), 125.33 (s), 115.54 (s); 31P NMR (162 MHz, C6D6) δ: -12.8 (s). Anal. calcd for C38H32Cl4N2OP2Ti2: C 54.85, H 3.88, N 3.37; found C 54.98, H 3.94, N 3.42.
Ethylene polymerization experiments were carried out in a 150 mL of autoclave (150 mL scale stainless steel) equipped with a stirrer, an external circulation bath and a thermocouple to measure the reaction temperature. The polymerization conditions have listed in Table 2. The general operation steps are as follows. A proper amount of toluene and MAO were added into the autoclave. Thereafter a toluene solution of prepared catalyst was added into the autoclave, and the reaction apparatus was immediately pressurised to setting pressure using ethylene gas and keep stated pressure by gas flowmeter. The polymerization was terminated by adding a small volume of EtOH. Then the mixture was poured into EtOH (100 mL), and the polymer was isolated by filtering. The resultant polymers were dried until constant weight in vacuo.
Supporting Information 1H NMR, 13C NMR, 31P NMR spectra for all compounds and crystallographic data. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn/.
(a) Gan, W. ; Nie, W. L. ; Chen, Y. F. Chin. J. Org. Chem. 2009, 29, 1200 (in Chinese).
(甘伟, 聂万丽, 陈耀峰, 有机化学, 2009, 29, 1200. )
(b) Xu, S. S. ; Wang, B. Q. ; Yuan, F. ; He, S. S. ; Zhou, X. Z. ; Zou, F. L. ; Li, Y. Chin. J. Org. Chem. 2003, 23, 187 (in Chinese).
(徐善生, 王佰全, 袁方, 贺双胜, 周秀中, 邹丰楼, 李杨, 有机化学, 2003, 23, 187. )
(c) Resconi, L. ; Cavallo, L. ; Fait, A. ; Piemontesi, F. Chem. Rev. 2000, 100, 1253.
(d) Hessen, B. J. Catal. 2004, 213, 129.
(a) Kang, X. H. ; Zhou, G. L. ; Wang, X. B. ; Qu, J. P. ; Hou, Z. M. ; Luo, Y. Organometallics 2016, 35, 913.
(b) Valente, A. ; Mortreux, A. ; Visseaux, M. ; Zinck, P. Chem. Rev. 2013, 113, 3836.
(c) Resconi, L. ; Balboni, D. ; Baruzzi, G. ; Fiori, C. ; Guidotti, S. Organometallics 2000, 19, 420.
(d) Talja, M. ; Polamo, M. ; Leskela, M. J. Catal. 2008, 280, 102.
(a) Nikitin, S. V. ; Nikitin, V. V. ; Oleynik, I. I. ; Oleynik, I. V. ; Bagryanskaya, E. G. J. Catal. 2016, 423, 285.
(b) Srijumnong, S. ; Suttipitakwong, P. ; Jongsomjit, B. ; Praserthdam, P. J. Catal. 2008, 294, 1.
(c) Wang, Y. X. ; Zuo, M. H. ; Li, Y. S. Chin. J. Catal. 2015, 36, 657.
(d) Yan, Q. ; Yang, W. H. ; Chen, L. Q. ; Wang, L. ; Redshaw, C. ; Sun, W. H. J. Organomet. Chem. 2014, 753, 34.
(e) Nomura, K. ; Pengoubol, S. ; Apisuk, W. RSC Adv. 2016, 6, 16203.
(a) Hu, W. Q. ; Sun, X. L. ; Wang, C. ; Gao, Y. ; Tang, Y. ; Shi, L. P. ; Xia, W. ; Sun, J. ; Dai, H. L. ; Li, X. Q. ; Yao, X. L. ; Wang, X. R. Organometallics 2004, 23, 1684.
(b) Qi, C. H. ; Zhang, S. B. Appl. Organomet. Chem. 2006, 20, 70.
(c) Liu, K. ; Wu, Q. ; Gao, W. ; Mu Y. ; Ye, L. Eur. J. Inorg. Chem. 2011, 12, 1901.
(d) Nomura, K. ; Liu, J. Dalton Trans. 2011, 7666.
(a) Liu, Q. Y. ; Gao, R. ; Hou, J. X. ; Sun, W. H. Chin. J. Org. Chem. 2013, 33, 808 (in Chinese).
(刘清云, 高榕, 侯俊先, 孙文华, 有机化学, 2013, 33, 808. )
(b) Yan, Q. ; Yang, W. H. ; Chen, L. Q. ; Wang, L. ; Redshaw, C. ; Sun, W. H. J. Organomet. Chem. 2014, 753, 34.
(c) Su, B. Y. ; Jia, P. Y. ; Wang, Y. Z. ; Li, Y. N. ; Huang, H. ; Li, Q. D. Chin. J. Org. Chem. 2016, 36, 2344 (in Chinese).
(苏碧云, 郏佩瑜, 王彦昭, 李亚宁, 黄鹤, 李谦定, 有机化学, 2016, 36, 2344. )
(d) Stephan, D. W. ; Stewart, J. C. ; Guérin, F. ; Spence, R. E. v. H. ; Xu, W. ; Harrison, D. G. Organometallics 1999, 18, 1116.
(a) Stephan, D. W. Organometallics 2005, 24, 2548.
(b) Dehnicke, K. ; Krieger, M. ; Massa, W. Coord. Chem. Rev. 1999, 182, 19.
(c) Chen, J. J. ; Wang, T. S. ; Tang, Z. W. ; Xu, Y. B. ; Xu, l. ; Cao, M. S. ; Feng, Z, G. Acta Polym. Sin. 2017, (8), 1294 (in Chinese).
(陈建军, 王铁石, 唐正伟, 徐一兵, 徐林, 曹茂盛, 冯增国, 高分子学报, 2017, (8), 1294. )
(a) Hollink, E. ; Stewart, J. C. ; Wei, P. R. ; Stephan, D. W. Can. J. Chem. 2004, 82, 1304.
(b) Siemeling, U. ; Kö lling, L. ; Stammler, A. ; Stammler, H-G. J. Chem. Soc., Dalton Trans. 2002, 3277.
(c) Sarsfield, M. J. ; Said, M. ; Thornton-Pett, M. ; Gerrard, L. A. ; Bochmann, M. J. Chem. Soc., Dalton Trans. 2001, 822.
(c) Wang, T. S. ; Chen, J. J. ; Ye, L. ; Zhang, A. Y. ; Feng, Z, G. Chin. J. Org. Chem. 2018, 38, 1544 (in Chinese).
(王铁石, 陈建军, 叶霖, 张爱英, 冯增国, 有机化学, 2018, 38, 1544. )
(a) Xu, S. ; Feng, Z. F. ; Huang, J. L. J. Catal. 2006, 250, 35.
(b) Resconi, L. ; Balboni, D. ; Baruzzi, G. ; Fiori, C. ; Guidotti, S. Organometallics 2000, 19, 420.
(c) Zhang, Q. ; Tao, X. ; Gao, W. ; Song, T. T. ; Mu, Y. J. Org. Chem. 2016, 804, 18.
(a) Xu, S. ; Liang, C. C. ; Lv, Z. W. ; Zhu, Y. L. ; Zhang, C. ; Mi, P. K. Chin. J. Org. Chem. 2017, 37, 1284 (in Chinese).
(许胜, 梁春超, 吕中文, 朱玉玲, 张翠, 米普科, 有机化学, 2017, 37, 1284. )
(b) Ewen, J. A. ; Elder, M. J. ; Jones, R. L. ; Rheingold, A. L. ; Liable-Sands, L. M. ; Sommer, R. D. J. Am. Chem. Soc. 2001, 123, 4763.
(c) Yamazaki, H. ; Kimura, K. ; Nakano, M. ; Ushioda, T. Chem. Lett. 1999, 1311.
(d) Williams, L. A. ; Marks, T. J. Organometallics 2009, 28, 2053.
(a) Li, H. ; Stern, C. L. ; Marks, T. J. Macromolecules 2005, 38, 9015.
(b) Haak, R. M. ; Wezenberg, S. J. ; Kleij, A. W. Chem. Commun. 2010, 46, 2713.
(c) Delferro, M. ; Marks, T. J. Chem. Rev. 2011, 111, 2450.
(d) Li, H. ; Marks, T. J. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 15295.
(e) Liu, S. ; Motta, A. ; Delferro, M. ; Marks, T. J. J. Am. Chem. Soc. 2013, 135, 8830.
McInnis, J. P.; Delferro, M.; Marks, T. J. Acc. Chem. Res. 2014, 47, 2545. doi: 10.1021/ar5001633
(a) Guo, N. ; Li, L. T. ; Marks, T. J. J. Am. Chem. Soc. 2004, 126, 6542.
(b) Li, H. ; Li, L. ; Marks, T. J. ; Liable-Sands, L. ; Rheingold, A. L. J. Am. Chem. Soc. 2003, 125, 10788.
(c) Motta, A. ; Fragala, I. L. ; Marks, T. J. J. Am. Chem. Soc. 2009, 131, 3974.
(d) Li, H. ; Li, L. ; Marks, T. J. Angew. Chem., Int. Ed. 2004, 43, 4937.
(e) Li, L. ; Metz, M. V. ; Li, H. ; Chen, M. -C. ; Marks, T. J. ; Liable-Sands, L. ; Rheingold, A. L. J. Am. Chem. Soc. 2002, 124, 12725.
(a) Chahma, M. ; Myles, D. J. T. ; Hicks, R. G. Can. J. Chem. 2005, 83, 150.
(b) Stott, T. L. ; Wolf, M. O. J. Phys. Chem. B 2004, 108, 18815.
(c) Reiter, S. A. ; Nogai, S. D. ; Schmidbaur, H. Dalton Trans. 2005, 247.
(d) Sevcik, R. ; Necas, M. ; Novosad, Polyhedron 2003, 22, 1585.
Latham, I. A.; Leigh, G. J. J. Chem. Soc., Dalton Trans. 1986, 377.
Stephan, D. W.; Stewart, J. C.; Guérin, F.; Courtenay, S.; Kickham, J. E.; Hollink, E.; Beddie, C.; Hoskin, A. J.; Graham, T.; Wei, P.; Spence, R. E. v. H.; Xu, W.; Koch, L.; Gao, X.; Harrison, D. G. Organometallics 2003, 22, 1937. doi: 10.1021/om020954t
(a) Resconi, L. ; Cavallo, L. ; Fait, A. ; Piemontesi, F. Chem. Rev. 2000, 100, 1253.
(b) Jin, Y. L. ; Zhao, N. ; Cheng, R. H. ; Liu, B. P. CIESC J. 2017, 68, 485 (in Chinese).
(金玉龙, 赵柠, 程瑞华, 刘柏平, 化工学报, 2017, 68, 485. )
Chen J. X.; Huang Y. B.; Li Z. S.; Zhang Z. C.; Wei C. X.; Lan T. Y.; Zhang W. J. J. Mol. Catal. A: Chem. 2006, 259, 133. doi: 10.1016/j.molcata.2006.06.016
Xiao X. H.; Sun J. Q.; Li X.; Wang Y. G.; Schumann H. Eur. Polym. J. 2007, 43, 164. doi: 10.1016/j.molcata.2006.06.016

Figure 2 Mono- (Type A) and bisdiphenylphosphine (Type B) substituted pyridine, thiophene and furan with titanium halfmetallocene complexes

Scheme1 Synthesis of mono- and bimetallic heteoarylphos- phinimine Ti complexes 3a~3c and 6a~6c

Figure 3 Molecular structures of 3a, 3b and 3c, with thermal ellipsoids shown at the 50% probability level

Figure 4 Molecular structures of 6a, 6b and 6c, with thermal ellipsoids shown at the 50% probability level Hydrogen atoms have been omitted for clarity
Table 1. Selected bond distances (Å) and angles (°) for complexes 3a~3c and 6a~6ca
| 2-(Ph2PNTiCl2Cp)C5H4N (3a) | |||
| Ti(1)—N(1) | 1.779(3) | Cl(2)—Ti(1)—Cl(1) | 102.17(6) |
| P(1)—N(1) | 1.594(3) | P1—N(1)—Ti(1) | 167.10(18) |
| P(1)—C(6) | 1.809(3) | N(2)—C(6)—P(1) | 116.9(2) |
| P(1)—Cl(2) | 1.801(3) | N(1)—P(1)—C(6) | 111.70(15) |
| P(1)—C1(8) | 1.807(3) | N(1)—P(1)—C(12) | 113.31(15) |
| Ti(1)—Cl(1) | 2.3033(13) | N(1)—P(1)—C(18) | 109.84(15) |
| Ti(1)—Cl(2) | 2.2955(13) | N(1)—Ti(1)—Cpa | 120.329(106) |
| 2, 6-(Ph2PNTiCl2Cp)2C5H3N (6a) | |||
| Ti(1)—N(1) | 1.784(3) | Cl(2)—Ti(1)—Cl(1) | 101.52(4) |
| P(2)—N(1) | 1.578(3) | P(2)—N(1)—Ti(1) | 161.75(19) |
| P(2)—C6 | 1.800(3) | N(2)—C(12)—P(2) | 114.7(2) |
| P(2)—C12 | 1.821(3) | N(1)—P(2)—C(6) | 114.76(15) |
| P(2)—C15 | 1.800(3) | N(1)—P(2)—C(12) | 108.69(15) |
| Ti(1)—Cl(1) | 2.3014(11) | N(1)—P(2)—C(15) | 111.07(15) |
| Ti(1)—Cl(2) | 2.2900(11) | N1—Ti(1)—Cpa | 117.769(93) |
| 2-(Ph2PNTiCl2Cp)2C4H3S (3b) | |||
| Ti(1)—N(1) | 1.775(4) | Cl(2)—Ti(1)—Cl(1) | 102.55(6) |
| P1—N(1) | 1.590(4) | P1—N(1)—Ti(1) | 164.3(2) |
| P(1)—C4 | 1.781(4) | S(1)—C(4)—P(1) | 122.0(2) |
| P(1)—C5 | 1.800(4) | N(1)—P(1)—C(4) | 114.45(19) |
| P(1)—C11 | 1.795(4) | N(1)—P(1)—C(5) | 111.60(19) |
| Ti(1)—Cl(1) | 2.3184(14) | N(1)—P(1)—C(11) | 110.78(19) |
| Ti(1)—Cl(2) | 2.2963(15) | N1—Ti(1)—Cpa | 120.213(132) |
| 2, 5-(Ph2PNTiCl2Cp)2C4H2S (6b) | |||
| Ti(1)—N(1) | 1.771(4) | Cl(2)—Ti(1)—Cl(1) | 101.65(7) |
| P1—N(1) | 1.596(4) | P(1)—N(1)—Ti(1) | 162.2(2) |
| P(1)—C1 | 1.800(4) | S(1)—C(13)—P(1) | 118.8(2) |
| P(1)—C7 | 1.789(4) | N(1)—P(1)—C(1) | 113.97(19) |
| P(1)—C13 | 1.792(4) | N(1)—P(1)—C(7) | 109.92(19) |
| Ti(1)—Cl(1) | 2.3033(14) | N(1)—P(1)—C(13) | 109.14(19) |
| Ti(1)—Cl(2) | 2.2906(17) | N1—Ti(1)—Cpa | 118.233(119) |
| 2-(Ph2PNTiCl2Cp)C4H3O (3c) | |||
| Ti(1)—N(1) | 1.778(2) | Cl(1)—Ti(1)—Cl(2) | 101.98(4) |
| P1—N(1) | 1.587(2) | P(1)—N(1)—Ti(1) | 162.99(15) |
| P(1)—C(1) | 1.782(3) | O(1)—C(1)—P(1) | 116.09(19) |
| P(1)—C(5) | 1.791(3) | N(1)—P(1)—C(1) | 114.22(12) |
| P(1)—C(11) | 1.801(3) | N(1)—P(1)—C(5) | 111.70(12) |
| Ti(1)—Cl(1) | 2.3021(10) | N(1)—P(1)—C1(1) | 111.84(12) |
| Ti(1)—Cl(2) | 2.3093(9) | N1—Ti(1)—Cpa | 119.630(73) |
| 2, 5-(Ph2PNTiCl2Cp)2C4H2O (6c) | |||
| Ti(1)—N(1) | 1.791(2) | Cl(2)—Ti(1)—Cl(1) | 101.40(4) |
| P1—N(1) | 1.576(2) | P(1)—N(1)—Ti(1) | 161.91(15) |
| P(1)—C(1) | 1.793(3) | O(1)—C(13)—P(1) | 118.47(17) |
| P(1)—C(7) | 1.801(3) | N(1)—P(1)—C(1) | 117.63(12) |
| P(1)—C(13) | 1.792(3) | N(1)—P(1)—C(7) | 110.74(12) |
| Ti(1)—Cl(1) | 2.3042(9) | N(1)—P(1)—C(13) | 107.73(12) |
| Ti(1)—Cl(2) | 2.2922(10) | N1—Ti(1)—Cpa | 118.739(76) |
| a N—Ti—Cp angles refer to the angle at Ti between N and the centroid of the five η5-carbons of the Cp-type ligands. | |||
 下载: 导出CSV
下载: 导出CSV
Table 2. Ethylene polymerization conditions and characterizationa
| Entry | Cat. | Cat./μmol | T/℃ | Time/min | Yield/g | Activityb×106 | Tmc/℃ | Mnd×104 | Mwd×104 | PDId |
| 1 | 3a | 20 | 50 | 15 | 2.15 | 0.43 | 142.7 | 9.44 | 68.10 | 7.2 |
| 2 | 3b | 20 | 50 | 15 | 4.25 | 0.85 | 141.5 | 7.04 | 58.19 | 8.3 |
| 3 | 3c | 20 | 50 | 15 | 3.16 | 0.63 | 140.7 | 5.86 | 52.26 | 8.9 |
| 4 | 6a | 10 | 50 | 15 | 3.21 | 1.28 | 140.5 | 5.60 | 47.42 | 8.5 |
| 5 | 6b | 10 | 50 | 15 | 4.69 | 1.88 | 140.5 | 10.26 | 48.31 | 4.7 |
| 6 | 6c | 10 | 50 | 15 | 3.15 | 1.26 | 143.1 | 5.59 | 66.97 | 12.0 |
| 7 | 6b | 10 | 40 | 15 | 3.45 | 1.38 | 143.6 | 8.52 | 121.7 | 14.3 |
| 8 | 6b | 10 | 30 | 15 | 3.31 | 1.32 | 145.3 | 10.75 | 179.7 | 16.7 |
| 9 | 6b | 10 | 30 | 5 | 2.42 | 2.90 | 144.1 | 9.59 | 172.3 | 18.0 |
| a Polymerization conditions: 150 mL stainless steel autoclave, 100 mL of toluene, under 0.5 MPa. b Activity in units of g (mol•cat.)-1 h-1. c Melting temperature was determined by differential scanning of calorimetry (DSC). d Determined by GPC at 160 ℃ in trichlorobenzene using polystyrene standards, given in units of g• mol-1. | ||||||||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们

 下载:
下载: