Citation:
Jiahao Huang, Yongfeng Lan, Lifa Chen, Xiaoxuan Guan, Rong Liang, Jiatong Liang, Ming Gao, Jialiang Gan, Yun Guo, Sen Zhang. Living therapeutics: Colorectal related diseases precision therapy with engineered organisms[J]. Chinese Chemical Letters,
2026, 37(6): 112151.
doi:
10.1016/j.cclet.2025.112151
Living therapeutics: Colorectal related diseases precision therapy with engineered organisms
English
Living therapeutics: Colorectal related diseases precision therapy with engineered organisms
zs0771@126.com (S. Zhang). 1 These authors contributed equally to this work.
Received Date:
17 June 2025 Accepted Date:
21 November 2025 Revised Date:
20 November 2025 Available Online:
15 June 2026
Abstract:
Colorectal related diseases are generally benign and malignant diseases that occur within the colon, causing psychological disorders, nutrient absorption disorders, anemia, and life threatening conditions. The current treatment strategies for colorectal related diseases have been seriously hampered by unpredictable behaviors, safety risks and limited efficacy. Recent advances in the fields of synthetic biology and materials fabrication have enabled the development of engineered organisms with great controllability, targeted delivery capabilities, and high safety and efficacy for their therapy. In this review, we first analyze the mechanisms underlying the occurrence and development of colorectal related disease. Subsequently, we delved into the latest developments in the application of engineered organisms in the treatment of colorectal diseases, covering the potential regulatory mechanisms, and the exploration of clinical feasibility. Finally, we discuss the key challenges and future perspectives of this biotherapeutic approach, with a focus on achieving precise targeted therapy through the engineering design of organisms. This review aims to provide the valuable insights into the development of precision therapies for colorectal related diseases, and lay the foundation for their clinical management.
Colorectal related diseases refer to various conditions affecting the colon and rectum, encompassing inflammatory disorders, benign tumors, and malignant tumors. Common examples include colorectal cancer (CRC), inflammatory bowel disease (IBD), and radiation-induced colorectal fibrosis, all of which pose serious threats to human health. For example, CRC is a malignant tumor with extremely high incidence and mortality rates worldwide [1,2]. Early symptoms are often subtle, and may delay diagnosis. As the condition progresses, symptoms such as bloody stools, abdominal pain, and changes in bowel habits may appear [3]. Without timely intervention, cancer cells may metastasize to organs such as the liver and lung, posing a life threatening risk [4]. And IBD is characterized by chronic inflammation of the intestines [5,6], and its pathological mechanisms involve abnormal activation of the immune system, and disruption of intestinal barrier function [7-9]. Chronic inflammation always leads to damage of intestinal tissues, and triggers symptoms such as diarrhea and abdominal pain [10]. It may also result in electrolyte imbalances and malnutrition [11]. In addition, IBD is characterized by recurrent episodes and chronicity, often leading to complications such as intestinal obstruction and perforation. Notably, the chronic inflammatory state of IBD may promote CRC development through the inflammation-dysplasia-carcinoma sequence, significantly increasing patients’ cancer risk [12,13].
The effective therapeutic strategies are crucial for the clinical prognosis of patients with colorectal related diseases. Following early intervention of CRC, the combined approaches such as surgery with chemoradiotherapy can eliminate lesions, suppress metastasis, and improve survival rates. Timely and standardized treatment for IBD patients can alleviate symptoms, reduce complications, and lower the risk of malignant transformation [14]. However, traditional treatments (surgery, radiotherapy and chemotherapy, anti-inflammatory drugs, etc.) have limitations. For example, surgery involves significant trauma and slow recovery. Radiotherapy and chemotherapy are highly toxic, and prone to causing bone marrow suppression. Drugs lack specificity, leading to drug resistance with long term use, and may also increase the risk of infection and organ damage [15,16].
With the development of nanobiotechnology, novel therapeutic strategies such as targeted drugs [17,18], biologics [19] and cell therapy [20] can address the limitations of conventional treatments. Biological therapies for colorectal related diseases utilize live organisms such as probiotics or their metabolites to modulate the gut microbiota, strengthen the intestinal barrier, suppress pathogenic bacteria, reduce inflammation, and promote epithelial repair [21]. It can also activate immune cells to maintain intestinal immune homeostasis [22,23]. Research has found that short chain fatty acids (such as butyrate) produced by the organisms can inhibit tumor cell proliferation, and induce apoptosis [24,25]. Biological therapies are more precise than traditional treatments. They can offer superior effects in regulating immunity, suppressing tumors, or improving inflammation, while also demonstrate better safety and tolerability.
It is worth noting that the biological therapies face limitations in targeting precision, immune responses, and delivery methods. Natural organisms also exhibit poor stability and controllability due to individual variations in gut microbiota and intestinal environment, making clinical treatment challenging. To address these issues, engineered organisms have emerged. The engineered organisms created through technologies like gene editing, nanotechnology combination, and biomimetic technology [26,27], hold promise in enhancing treatment precision, efficacy and safety, offering novel methods for managing colorectal related diseases.
2.
Mechanisms in colorectal related diseases
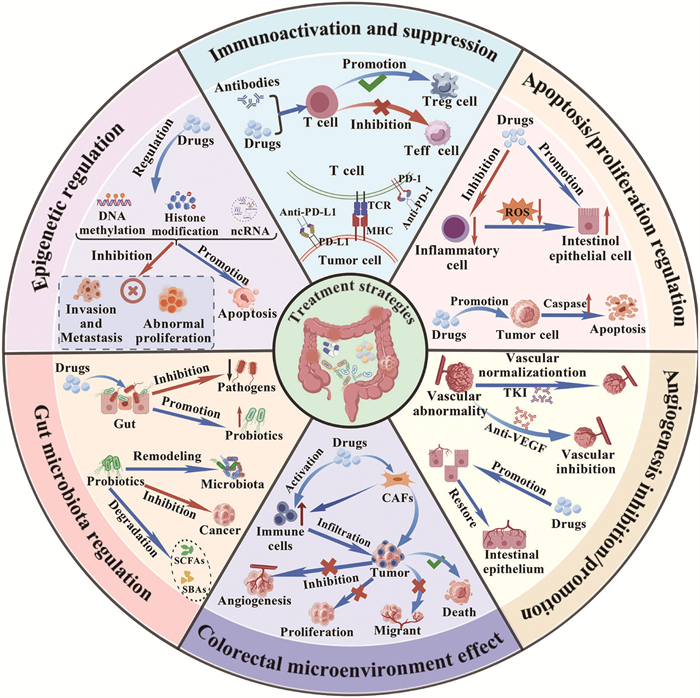
The occurrence and development of colorectal related diseases is always associated with various mechanisms involving immune activation/suppression, apoptosis/proliferation regulation, angiogenesis, gut microbiota homeostasis, and epigenetic modifications. Corresponding therapeutic strategies can be devised to target each of these mechanisms (Fig. 1).
Figure 1
Figure 1.
The main therapeutic mechanism for colorectal related diseases: immunoactivation and suppression, apoptosis/proliferation regulation, angiogenesis inhibition/promotion, colorectal microenvironmental effect, gut microbiota regulation, and epigenetic regulation.
The immune system plays a crucial role in the development and progression of colorectal related diseases. Under healthy conditions, immune cells can recognize and eliminate abnormal cells or pathogens. However, in CRC, tumor cells evade immune surveillance through multiple mechanisms, one of which is the abnormal activation of immune checkpoint pathways. The binding of programmed death 1 (PD-1) to programmed death ligand 1 (PD-L1) normally functions as an immunosuppressive signal, preventing excessive immune responses and damage to self-tissues [28]. However, tumor cells achieve immune escape by highly expressing PD-L1, which binds to PD-1 of T cell surface, and subsequently suppresses T cell activity. Immune checkpoint inhibitors (such as PD-1/PD-L1 inhibitors) can block this interaction, and restore T cells antitumor function, demonstrating the significant efficacy in patients with microsatellite instability-high (MSI-H) or mismatch repair deficiency (dMMR) CRC [29,30].
In addition, the tumor microenvironment (TME), serving as the complex ecosystem in which tumor cells thrive, comprises immune cells, fibroblasts, extracellular matrix (ECM), and various cytokines, playing a pivotal regulatory role in the immune response process [31]. Immunotherapy not only directly activates T cells and natural killer (NK) cells to kill tumors, but also systematically reshapes the immune state of TME [32,33]. Following immunactivation, infiltrating T cells and NK cells release interferon-γ (IFN-γ), which downregulates PD-L1 expression on tumor cells, and create a positive feedback loop to amplify the immune response [34]. Simultaneously, they induce tumor associated macrophages (TAMs) to shift from M2 to M1 polarization, enhancing their antigen presentation and pro-inflammatory functions [35]. Similarly, factors such as tumor necrosis factor-alpha (TNF-α) secreted by activated immune cells can influence tumor associated fibroblasts (CAFs), promoting their transition from a tumorigenic phenotype to a relatively quiescent state. This reduces ECM deposition, and decreases secretion of immunosuppressive factors like transforming growth factor-beta (TGF-β), thereby weakening the immunosuppressive functions of regulatory T cells (Tregs) and myeloid derived suppressor cells (MDSCs). This process involves upregulation of major histocompatibility complex (MHC)-Ⅰ/Ⅱ molecules, enhanced co-stimulatory signaling, and formation of inflammatory cytokine gradients, collectively boosting tumor immunogenicity, and promoting clearance effects [36]. Furthermore, targeted modulation of CAFs activity can alter ECM composition, and inhibit tumor migration and metastasis [37-39].
In IBD, aberrant activation of the immune system is a central pathological feature. The immune system incorrectly recognizes intestinal mucosal tissues as foreign antigens, triggering a sustained inflammatory response [40]. The central goal of treatment is to suppress the overactive immune response, and restore immune homeostasis. Glucocorticoids, as classical immunosuppressions, alleviate intestinal inflammation by inhibiting the activation and migration of inflammatory cells, and reducing the release of pro-inflammatory cytokines (e.g., TNF-α, interleukin (IL)-1β) [41]. In addition, biological agents (e.g., anti-TNF-α monoclonal antibodies) inhibit the inflammatory cascade response by specifically binding to TNF-α, and blocking its interaction with the receptor, which significantly improve the clinical symptoms and intestinal pathological damage in patients [42]. In recent years, targeted therapies against other inflammatory mediators (IL-12/IL-23, integrins, etc.) have also made important progress, providing more options for the treatment of IBD [43,44].
2.2
Apoptosis/proliferation regulation
Apoptosis is a process of programmed cell death that plays multiple roles in the treatment of colorectal related diseases. Its regulatory mechanisms involve both intrinsic and extrinsic apoptotic pathways as well as the autophagy process [45-47]. By precisely intervening these pathways, effective strategies can be provided for the treatment of colorectal related diseases.
In the treatment of CRC, induction of apoptosis in tumor cells is an important strategy. Chemotherapeutic agents (e.g., fluorouracil, oxaliplatin) disrupt cell cycle progression by interfering with DNA synthesis and replication in tumor cells, which in turn activate apoptotic signaling pathways. This key regulatory proteins, such as tumor protein p53, can upregulate the expression of pro-apoptotic proteins (e.g., Bcl-2 associated X protein (Bax)), while simultaneously suppress the function of anti-apoptotic proteins (e.g., B-cell lymphoma-2 (Bcl-2)) [48]. This process directly leads to excessive production of reactive oxygen species (ROS) within the cell, and alterations in the mitochondrial membrane potential (MMP). The collapse of MMP constitutes the decisive event in the intrinsic pathway. It results in mitochondrial outer membrane permeabilisation (MOMP), causing cytochrome C to be released into the cytoplasm. Within the cytoplasm, cytochrome C assembles with the apoptosis related protein activator-1 (Apaf-1) and procaspase-9 to form a crucial protein complex known as the “apoptosome”. The formation of the apoptosome activates the caspase-9, thereby initiating the subsequent caspase cascade reaction [49,50].
Some targeted drugs can induce tumor cell apoptosis through specific molecular targets. For example, drugs targeting Bcl-2 family proteins can block the apoptosis-suppressing function of Bcl-2, and promote apoptosis of tumor cells [51]. The Bcl-2 family proteins play a key role in the regulation of mitochondria-dependent apoptotic pathway, and their imbalance is an important mechanism for tumor cells to escape from apoptosis [52]. In CRC, overexpression or mutation of Bcl-2 family proteins can disrupt endogenous pathways, making them a strategy to restore apoptosis sensitivity. Additionally, the extrinsic apoptosis pathway (death receptor pathway) also serves as a crucial mechanism for inducing tumor cell apoptosis [53]. The extrinsic pathway is initiated by extracellular death ligands (such as Fas ligand (FasL), TNF-α, tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)) binding to cell surface death receptors like Fas/CD95 or tumor necrosis factor receptor 1 (TNFR1). Upon receptor-ligand binding, trimerization occurs, and intracellular adaptor proteins are recruited, forming the death inducing signaling complex (DISC). The DISC recruits and activates the caspase cascade, leading to apoptosis [54]. In CRC, the death receptor pathway is often inactivated due to receptor downregulation or mutation. Therefore, activating the death receptor pathway represents a strategy to induce tumor cell apoptosis while minimizing toxicity to normal cells. Additionally, autophagy, as a process of cellular self-degradation, is closely associated with apoptosis [55]. Modulation of autophagy related proteins (e.g., light chain 3-Ⅱ (LC3-Ⅱ), Beclin-1) regulates tumor cell fate decisions between survival and apoptosis [56]. In IBD, aberrant apoptosis of intestinal epithelial cells compromises the mucosal barrier integrity, exacerbating inflammatory responses. The therapeutic goal is to restore intestinal epithelial cell homeostasis by regulating apoptosis [57]. Anti-apoptotic drugs (e.g., growth factors) promote epithelial cell repair and regeneration, while pro-apoptotic drugs (e.g., TNF-α inhibitors) reduce the survival of inflammatory cells, thereby alleviating inflammation. Thus, targeting apoptosis/proliferation regulation was an effective strategy in the treatment of colorectal related diseases.
2.3
Angiogenesis inhibition/promotion
In CRC, tumor growth and metastasis rely on neovascularization to supply nutrients and oxygen. Malignant cells secrete pro-angiogenic factors such as vascular endothelial growth factor (VEGF), which stimulates endothelial cell proliferation, migration, and lumen formation, thereby promoting angiogenesis [58]. Antiangiogenic therapy is a key strategy to inhibit tumor growth. For example, bevacizumab, an anti-VEGF monoclonal antibody, specifically binds to VEGF, and blocks the binding to the vascular endothelial cell surface receptor (VEGFR), thereby inhibiting the proliferation and angiogenesis of vascular endothelial cells, cutting off the blood supply to tumors, restricting the nutrient acquisition of the tumor cells, inhibiting their growth, and inducing apoptosis [59].
In addition, small molecule tyrosine kinase inhibitors (TKIs) exert dual anti-angiogenic effects by targeting multiple pro-angiogenic receptors (including VEGFR, platelet-derived growth factor receptor (PDGFR) and fibroblast growth factor receptor (FGFR)), thereby suppressing downstream signaling cascades. This mechanism not only inhibits neovascularization, but also normalizes the aberrant architecture of existing tumor vasculature [60]. This effect helps to enhance the delivery efficiency of chemotherapeutic drugs, and improve the therapeutic effect [61]. In colorectal polyps, although the degree of angiogenesis is much lower than that of malignant tumors, persistent abnormal angiogenesis provides the necessary nutritional support for polyp growth. Drugs that inhibit angiogenesis can inhibit further polyp enlargement, and reduce the risk of malignancy by reducing neovascularization within the polyp, and limiting its nutrient supply. Therefore, anti-angiogenic therapy not only plays an important role in CRC, but also has potential application in the intervention of colorectal polyps.
However, In IBD, adequate angiogenesis is also involved in disease development, and promoted angiogenesis can attenuate inflammatory response and tissue injury, and promote tissue repair. Research indicated that endothelial dysfunction occurred in IBD, which compromised vascular barrier integrity, and exacerbated leukocyte infiltration and inflammatory responses. It was also found that SAR247799, a sphingosine-1-phosphate receptor 1 (S1PR1)-biased agonist, specifically activated the downstream Gi signalling pathway. This activation helped restore the integrity of the intestinal vascular endothelial barrier, thereby reducing abnormal permeation of inflammatory cells, and alleviating disease progression [62].
From the above, treating different colorectal related diseases separately with angiogenesis inhibition/promotion therapy is also a therapeutic option that can be considered.
2.4
Gut microbiota regulation
The gut microbiota refers to the entire community of microorganisms colonising the human gastrointestinal tract, encompassing bacteria, fungi, viruses, archaea and other microorganisms, with bacteria constituting the predominant component.
The gut microbiota plays a pivotal role in the pathogenesis of CRC and IBD. In IBD patients, significant microbial dysbiosis is observed, characterized by reduced abundance of commensal probiotics (e.g., Bifidobacterium, Lactobacillus) and enrichment of pathobionts (e.g., Escherichia coli, Clostridium) [63,64]. Probiotic therapy, as an effective strategy to regulate intestinal flora, can effectively improve intestinal microecological balance by supplementing beneficial flora [65,66]. These beneficial bacteria inhibit the over proliferation of pathogenic bacteria through mechanisms such as competition for nutrient substrates, and secretion of antimicrobial peptides and short-chain fatty acids (SCFAs), and at the same time play an anti-inflammatory role by regulating the immune response of the intestinal mucosa, enhancing the epithelial barrier function, and inhibiting the expression of pro-inflammatory factors (e.g., TNF-α, IL-6) [67,68]. Prebiotics (e.g., oligofructose, inulin), as exclusive metabolic substrates for beneficial bacteria, can selectively promote their proliferation, and further optimize the intestinal flora structure [69]. During the CRC process, the gut microbiota is closely associated with tumor initiation and progression [70,71]. Metabolites derived from specific gut bacteria (e.g., Fusobacterium nucleatum, enterotoxigenic Bacteroides fragilis), including secondary bile acids and hydrogen sulfide, exhibit procarcinogenic properties by inducing DNA damage and chronic inflammation, thereby promoting tumorigenesis [72,73]. In contrast, specific probiotics, such as Lactobacillus and Bifidobacterium, can exert anticancer effects through mechanisms such as degradation of carcinogens, modulation of the immune microenvironment, and induction of apoptosis in tumor cells [74-76].
In addition, fecal microbiota transplantation (FMT), as an emerging therapeutic tool [77-79], has shown some efficacy in refractory IBD. Some CRC patients associated with dysbiosis by FMT from a healthy donor have confirmed the possibility of re-establishing the normal ecology of the flora [80]. Although FMT shows promise in reshaping the gut microbiota, and treating refractory IBD and CRC, its clinical application remains constrained by significant long term safety risks and unpredictable microbial interactions. Recent studies reveal that transplantation may induce “regional microbial mismatch”, where colonic donor anaerobic bacteria abnormally colonize the small intestine. This could persistently alter local metabolic profiles and energy balance, and reshape gene expression in intestinal tissues, thereby exerting unforeseeable long term effects on host metabolism and immunity [81]. Furthermore, the efficacy of FMT exhibits significant individual variation, and lacks reliable predictive biomarkers, with its success depending more on the recipient’s initial gut microbiota state than on the donor itself. In specific, FMT may simultaneously transplant entire microbial communities including the virome and microbiome. The intricate and unpredictable interactions between these communities and the host immune system remain largely unclear. Combined with the lack of standardized technical procedures, these factors collectively pose potential unknown risks [82].
Although probiotics, prebiotics, and FMT hold therapeutic potentials for IBD and CRC through gut microbiota modulation, their safety profiles require careful consideration. Specifically, FMT presents long term safety risks, unpredictable microbial interactions, and variable individual responses, while the application of probiotics and prebiotics must be evaluated in the context of specific disease states to assess potential implications.
2.5
Epigenetic regulation
Epigenetic regulation also plays an important role in colorectal related diseases therapy. It is mainly achieved by demethylating drugs and histone deacetylase inhibitors (HDAC inhibitors) [83,84]. DNA methylation is a common epigenetic modification, and methylation inactivation of oncogenes is an important mechanism in CRC [85,86]. DNA methylation is catalyzed by DNA methyltransferases (DNMTs), which transfer the methyl group from S-adenosylmethionine to the 5′-carbon position of cytosine within CpG islands, forming 5-methylcytosine. In CRC, abnormal hypermethylation occurs in CpG islands within tumor suppressor gene promoter regions. This recruits methyl-CpG-binding domain proteins (MBDs) and histone deacetylases (HDACs), forming compact chromatin structures that cause transcriptional silencing. Consequently, these genes cannot exert their tumor-suppressing functions. Demethylating agents (e.g., decitabine) bind to DNMTs, promoting their degradation, and enabling both passive and active DNA demethylation. This reactivates the aforementioned tumor suppressor genes, inducing G2/M phase cell cycle arrest and apoptosis, thereby inhibiting tumor cell growth [87].
Histone modification is also an important aspect of epigenetic regulation. Altered histone acetylation levels affect chromatin structure and gene accessibility [88]. HDACs remove acetyl groups from histone lysine ε-amino groups, restoring their positive charge, and enhancing affinity for negatively charged DNA. This leads to chromatin condensation, and restricts transcription factor binding. In CRC, HDAC overexpression silences tumor suppressor genes and differentiation related genes. HDAC inhibitors block deacetylation by chelating zinc ions from the HDAC active site, leading to hyperacetylation of histones and chromatin relaxation. This promotes genes expression, including genes potentially involved in apoptosis and cell cycle regulation, thereby altering tumor cell biological behaviors [89,90]. In addition, non-coding RNAs such as micro RNAs (miRNAs) are also involved in epigenetic regulation [91-93] Certain miRNAs are abnormally expressed in CRC, which can inhibit translation or promote degradation by complementary pairing with target mRNAs to regulate the expression of tumor related genes, affecting the proliferation, apoptosis, and metastasis of tumor cells, and thus becoming a potential new target for CRC treatment [94,95].
Epigenetic regulation holds significant therapeutic value in colorectal related diseases, primarily mediated through three distinct mechanisms: demethylating agents reactivate tumor suppressor genes by degrading DNMTs. HDAC inhibitors modulate gene expression by suppressing HDAC activity, and promoting histone hyperacetylation. And non-coding RNAs such as microRNAs participate in epigenetic regulation by controlling tumor related gene expression. Together, these mechanisms offer potential therapeutic avenues for colorectal related disease management.
3.
Current therapeutic strategies
Colorectal related diseases represent highly prevalent gastrointestinal disorders worldwide, with persistently increasing incidence, a trend toward younger onset, and limited efficacy of current therapies, making them a critical challenge in both clinical and basic research. Current treatment modalities, including conventional radiotherapy and chemotherapy, immune checkpoint inhibitors, and biological agents, can control disease progression at certain stages but are constrained by limited efficacy, high drug resistance, inadequate targeting, and suboptimal safety profiles.
CRC is the third most common malignant tumor in the world, with the incidence rate increasing year by year and tends to be younger [96,97]. Its early symptoms are insidious, and systemic manifestations such as anemia and weight loss occur in the late stage. Although traditional surgery, radiotherapy and chemotherapy can reduce the tumor load, they can cause damages to normal tissues [98]. Significant progress has been made in both the optimization of comprehensive treatment strategies, and the development of new targeted drugs, bringing better survival benefits and better quality of life for patients. In the treatment of metastatic CRC, immune checkpoint inhibitors have become an important means of first- and backline therapy for patients with mismatch repair defects or highly unstable microsatellites, and the emergence of new targets and drugs has further improved the efficacy [99]. However, immune checkpoint inhibitors are effective in only 5% of MSI-H patients, and 95% of MSS patients have limited benefit [100]. And targeted therapies also face drug resistance problems, which limit their long term efficacy.
IBD is a group of chronic nonspecific inflammatory diseases of the gastrointestinal tract of unknown etiology with recurrent episodes, in which patients suffer from frequent diarrhea, severe abdominal pain, and other symptoms [101]. And in severe cases, they can lead to complications such as intestinal obstruction, abscesses, malnutrition, and so on, which not only pose a challenge in treatment, and reduce the quality of life of the patients, but also cause a heavy economic burden [102]. Although traditional IBD therapeutic agents such as 5-aminosalicylic acid and glucocorticoids can relieve symptoms, their application is limited due to their nonspecific anti-inflammatory effects and potentially serious side effects [103,104]. In recent years, biological agents such as TNF-α inhibitors, IL antagonists, and integrin receptor antagonists have been used in IBD treatment, which have overcome the limitations of conventional therapy to some extent [105-107]. However, these therapies have limited efficacy and significant side effects, and they can even lead to serious complications [108,109]. In the face of these challenges, there is an urgent need to develop safer and more effective drugs and methods for the management of IBD.
Although some progress has been made in the current treatment of colorectal related diseases including CRC and IBD, existing treatments still face challenges to be addressed in terms of drug resistance, precision and safety (Fig. 2). In recent years, with advances in biosynthetic technology and a deeper understanding of living organisms, there has been a shift toward the study of engineered organisms to overcome these obstacles. For example, by genetic modification, engineered organisms can specifically recognize and colonize the lesion site, release therapeutic drugs, or act directly to achieve precise treatment and reduce the impact on normal tissues [110]. More notably, engineered organisms give full play to the natural advantages of microorganisms to reshape the imbalanced intestinal micro-ecological environment by regulating the composition and function of the intestinal flora, and blocking the pathological development of diseases from the root process of disease development from the root. In CRC treatment, compared to the limitations of chemoradiotherapy, which causes intestinal mucosal damage, immunosuppression, and multi-organ toxicity, and traditional PD-1 inhibitors, which yield response rates below 15% in microsatellite-stable CRC, and frequently induce immune related adverse events, engineered biological agents demonstrate superior characteristics. They can target and colonize TME, enhancing disease control rates, and delivering therapeutic molecules. Side effects are more manageable, typically manifesting as mild hematologic reactions or low grade cytokine release syndrome [111-113]. In IBD treatment, nanozyme modified engineered probiotics can target inflammatory sites to scavenge ROS, and restore the intestinal barrier, demonstrating outstanding efficacy and favorable safety profiles in preclinical models [114-116]. Given their unique advantages, the therapeutic strategies of engineered organisms break through traditional limitations, emerging as a more promising treatment option in colorectal related diseases.
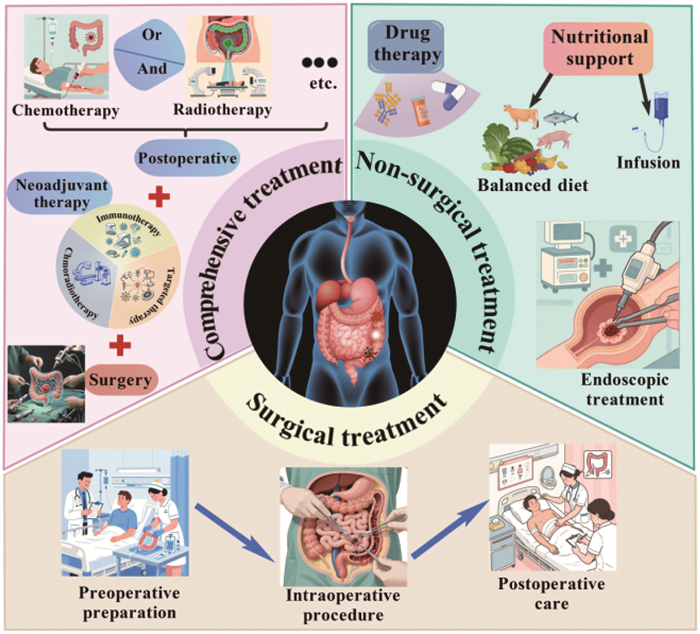
Figure 2
Figure 2.
Three primary therapeutic strategies for colorectal related diseases, namely non-surgical treatment, surgical intervention, and integrated management. Non-surgical approaches primarily involve conservative interventions, including medication, nutritional support, and endoscopic therapies, suitable for conditions such as active IBD. Surgical treatments focus on direct removal of lesions, addressing emergencies or refractory conditions such as CRC and intestinal obstruction. Integrated treatments pay attention to multidisciplinary collaboration, employing a “neoadjuvant therapy before surgery + surgical intervention + adjuvant therapy after surgery” model to enhance outcomes in complex cases like advanced CRC.
An engineered organism is an artificial biological system created through rational design, genetic circuit redesign, and the assembly of standardized biological parts to achieve predictable regulation or non-natural functions, characterized by its directed design, predictability, and functional programmability as opposed to the evolved traits of natural organisms. Generally, there are many defects in the application of natural organisms in biomedical fields. Their lack of specificity prevents them from pinpointing disease sites, and makes them difficult to accurately attack unhealthy cells. Influenced by the differences in individual intestinal flora, the therapeutic effect fluctuates greatly. And in the complex intestinal environment, the survival and function of natural organisms are limited, which greatly reduces the therapeutic efficacy. In clinical application, it is extremely difficult to accurately regulate the efficacy and range of their effects, which seriously limits the wide application of natural organisms in the treatment of colorectal related diseases.
The rapid advancement of engineered organisms as a new generation of therapeutic platforms stems from the emergence of cutting edge techniques such as synthetic biology and nanobiotechnology [117,118]. By strategically modifying, decorating, or reconstructing natural organisms, they can be endowed with the ability to precisely identify pathological sites, and efficiently intervene in disease progression. The recent design strategies of engineered organisms are displayed in Fig. 3. Leveraging their targeted controllability and programmable functionality, engineered organisms achieve multiple therapeutic objectives including precision delivery, immune modulation, and microenvironment remodeling, paving new technical pathways for personalized and precision medicine.
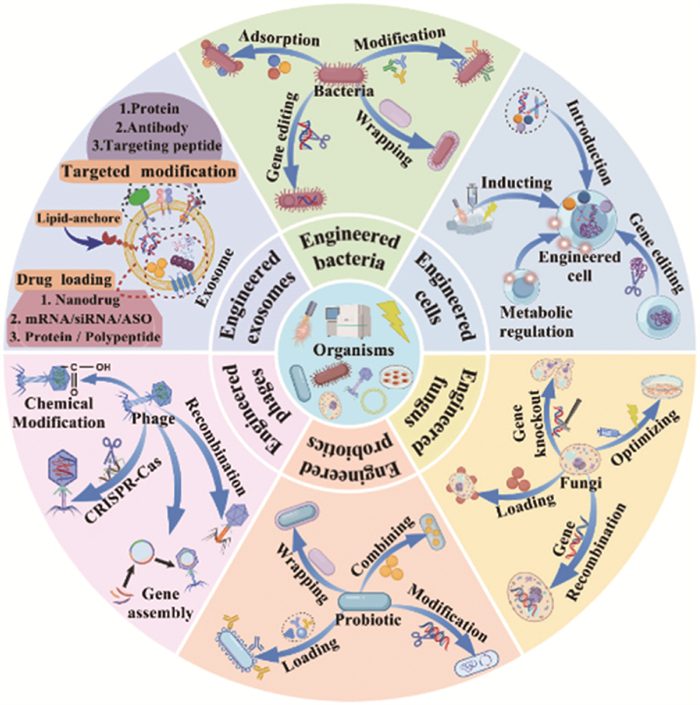
Figure 3
Figure 3.
The classification of engineered organisms, and their design strategies. The engineered organisms are mainly divided as engineered bacteria, cells, fungus, probiotics, phages and exosomes.
In recent years, a number of engineered organisms, have received extensive attention in the treatment of colorectal related diseases.The engineered organisms are mainly divided into engineered bacteria, fungi, cells, phages, probiotics and exosomes. In Table S1 (Supporting information), it illustrates the representative engineered organisms and their therapeutic mechanisms.
4.1
Engineered bacteria
Natural bacteria can regulate intestinal microecology, inhibit pathogenic bacteria, and enhance mucosal immunity in the prevention and treatment of colorectal related diseases. However, their colonization ability is weak, and their therapeutic effects are significantly affected by individual flora differences, potential risk of infection, and lack of precision. Therefore, in recent years, some researchers have been committed to improving the properties of natural bacteria through genetic modification and other methods, and endowing them with functions such as targeted colonization, drug responsive release, or immune microenvironmental modulation. Thus, they can play a better role in the prevention and treatment of colorectal related diseases [119].
For example, bacteria can be engineered as biosensors for the detection of CRC associated DNA mutations or tumor markers through engineering strategies. Hasty’s team developed a technology, which utilized Fusobacterium bezoar, a bacterium with the ability to transfer genes horizontally, genetically engineered to specifically recognize CRC associated mutant DNAs through the clustered regularly interspaced short palindromic repeats (CRISPR) system (e.g., KRASG12D). The modified bacterium carried a KRAS homology arm, distinguished between mutated and wild type DNA by CRISPR-Cas, and integrated only the mutated sequence to activate the kanamycin resistance gene as a detection signal. In vitro experiments demonstrated that this sensor could detect DNA from CRC cell lines and organoids. Following optimization, its sensitivity had increased approximately 10-fold, enabling analysis of crude lysates without the need of purification. In mouse experiments, rectally delivered engineered bacteria were shown to capture tumor DNA in vivo, and horizontal gene transfer events were detected in feces to distinguish tumor bearing from healthy mice [120]. Chen et al. studied the use of genetically engineered E. coli BL21 with overexpress catalase to alleviate tumor hypoxia, where PCN nanoparticles (PCN NPs) synthesized from the acoustic sensitizer tetrakis(4-carboxyphenyl)porphyrin (TCPP) and zirconium clusters (Zr6) were loaded on the surface of the bacteria by electrostatic adsorption to form the multifunctional biohybrid E.coli-pE@PCN. After intravenous injection, the hybrids exhibited excellent tumor targeting and penetration ability, not only continuously expressing catalase to improve TME, but also promoting the enrichment of acoustic sensitizers at the tumor site, which resulted in the generation of ROS triggered by ultrasound to kill tumor cells. It was further found that E.coli-pE@PCN based sonodynamic therapy (SDT) could not only effectively inhibit the growth of subcutaneous and in situ CRC, but also inhibit tumor recurrence and metastasis by releasing the immune adjuvant effects of tumor associated antigens and bacteria, activating dendritic cells (DCs) and cytotoxic T cells (CTLs), and inducing systematic anti-tumor immune responses. This dual mechanism of action strategy had been shown to be significantly superior to monotherapy in multiple mouse models [121].
In addition, Li et al. designed an orally available genetically engineered E. coli 1917. This bacterium, when stimulated by ultrasound, secreted enzymes containing the AH1 tumor rejecting epitope, the enzyme that produced the interferon gene stimulator (STING) agonist CDA, and a bacterial outer membrane vesicle (OMV) that targeted the microfollicular peptide Co1. In order to adapt them for oral administration, the bacteria were encapsulated using a polydopamine system (iPDA) to withstand the acidic environment of the stomach, improve their survival in vivo, and prolong their transit time in the intestine. Under the action of harmless ultrasound, the engineered OMV vaccine could be triggered to continuously secrete and traverse the intestinal epithelium to activate cyclic guanosine-adenosine synthetase (cGAS)-STING and Toll-like receptor 4 (TLR4) natural immune signaling pathways, which triggered a prolonged antigen specific immune response, and overcomed the immunosuppressive TME. In subcutaneous and in situ mouse CRC models, the system successfully inhibited tumor growth, and prolonged mouse survival without recurrence. Concurrently, polydopamine coating enabled resistance to gastric acidity, enhanced bacterial survival rates and intestinal transit time without significant safety concerns, demonstrating the potential to be a powerful oral vaccine system to improve colon cancer immunotherapy [122]. In addition, Zhang’s team successfully developed a L-cystine/cysteine dependent bacterial nanomedicine bio heterotrimer (DL@SFEc+) through a dual selection directed evolution technique. The researchers genetically modified E. coli to increase their L-cystine uptake capacity by 36-fold, and cysteine desulfurization enzyme activity by 23-fold, and combined these engineered bacteria with liposomes containing the vascular disrupting agent DMXAA to form a multifunctional therapeutic system. This biohybrid could specifically target TME, and the system exerted its anti-tumor effects through a dual mechanism. DMXAA disrupted tumor vasculature to block nutrient infiltration, and SFEc+ continuously depleted cystine (CySS) and cysteine (Cys) in TME to synergistically enhance metabolic therapeutic efficacy. The continuous depletion of CySS/Cys resulted in disruption of glutathione synthesis, and significant increased intracellular ROS and lipid peroxidation levels, inducing iron death and killing tumor cells. This biohybrid system significantly altered the tumor metabolic profile, involving oxidative stress and vascular injury related pathways. In colon cancer (CT26), pancreatic cancer (Panc-02) and spontaneous intestinal cancer (ApcMin/+) mouse models, it demonstrated highly efficient and low toxicity anti-tumor properties, almost completely inhibited the growth of spontaneous intestinal cancer with good biosafety [123].
In conclusion, the strategy of engineered bacteria enables precise targeting of tumors or lesions, reduces damage to normal tissues as well as unnecessary side effects, breaks through the bottleneck of insufficient targeting and metabolic compensation of traditional therapies, and provides a new solution to the challenge of treating colorectal related diseases. It is expected to further advance the research on their clinical translation.
4.2
Engineered cells
Cell therapy has demonstrated unique functional properties in colorectal related diseases, mainly including specific recognition of tumor antigens by immune cells, and activation of immune responses to kill tumor cells, or repair of damaged tissues and promotion of regeneration using stem cells. However, its application in colorectal related diseases is greatly limited by TME inhibition, low homing efficiency, and short survival time [124,125]. Therefore, some researchers have focused on engineering immune cells or stem cells to enhance their functional properties, and make them more useful in colorectal related diseases [126,127]. In particular, chimeric antigen receptor T cell immunotherapy (CAR-T) cell therapy genetically modified the patient’s T lymphocytes to enable them to target and remove cancer cells [128-130].
In recent years, some researchers have modified and enhanced CAR-T cell therapy to maximize its effectiveness, and minimize its harm (Fig. S1 in Supporting information) [131]. For example, Chen et al. constructed CAR-T cells against the CRC specific antigen carcinoembryonic antigen (CEA), and enabled them to autocrine PD-1-TREM2 single chain antibody fragments (scFv). It could act on both the immune checkpoint molecule PD-1/PD-L1 pathway, as well as the myeloid derived suppressor cells (MDSCs) and tumor related cells. Experimental results showed that it could effectively inhibit the activation of PD-1/PD-L1 pathway, block the binding of ligand to TREM2 receptors on MDSCs and TAMs, reduce the proportion of MDSCs and TAMs in TME, and enhance the effector function of T cells, thereby alleviating the immune resistance of TME. In a subcutaneous CRC mouse model, it had demonstrated more efficient tumor elimination than common CAR-T cells [132].
Müller et al. found that IBDs, with an imbalance between pro- and anti-inflammatory T cells in the lamina propria of the intestine, had difficulty in controlling inflammation efficiently by Tregs in the inflamed intestine. Previously, the team developed good manufacturing practice (GMP)-compliant in vitro expanded Tregs (IMP-Tregs) for autologous cell therapy, which showed promising results in a Phase Ⅰ trial. However, IMP-Tregs had limited intestinal homing ability and low expression of G protein coupled receptor 15 (GPR15). Therefore, researchers produced messenger ribonucleic acid (mRNA) for GPR15 using a GMP approved vector, and overexpressed it in IMP-Tregs by electroporation. Experiments showed that IMP-Tregs overexpressing GPR15 had enhanced dynamic adhesion to relevant adhesion molecules in vitro, and their homing in the intestinal lamina propria was significantly increased in a humanized mouse model. Meanwhile, co-culture experiments confirmed that overexpression of GPR15 did not affect the function of IMP-Tregs in inhibiting the proliferation of CD8+ T cells [133]. In addition, Wang et al. utilized genetically engineered modified and denucleated human mesenchymal stromal cells (MSCs) to achieve targeted delivery of therapeutic agents to diseased tissues. The research team first genetically modified MSCs to express chemoattractant receptors and endothelial cell binding molecules, and then denucleated them by density gradient centrifugation to construct a nucleated cell delivery system. The study found that MSCs did not proliferate or permanently colonize the host, but retained organelle maintenance functions, adhered to inflammatory endothelial cells through integrin regulation, and actively localized in the chemotactic gradient of diseased tissue. In a mouse model of acute inflammation and pancreatitis, systemic administration of enucleated cells expressing two chemokine receptors and endothelial adhesion molecules enhanced delivery of anti-inflammatory cytokines to diseased tissues, attenuated inflammation, and ameliorated pathology compared to unmodified stromal cells and bone marrow stromal cell derived exosomes. This delivery system combined the drug carrying properties of both cellular and cell free delivery systems, providing a new strategy for targeted therapies. It was expected to promote the clinical translation of precision medicine, or became the core platform for next generation targeted therapies [134].
In addition, CAR-T cell therapy has made great progress in hematological tumors (e.g., lymphoma, leukemia) [135], but when treating solid tumors, T cells are depleted and dysfunctional due to TME, resulting in poor efficacy. In order to enhance the metabolic resilience and anti-depletion ability of T cells, Zhao et al. genetically engineered CAR-T cells to secrete IL-10, which could maintain the structural and functional integrity of mitochondria, and enhance oxidative phosphorylation in a mitochondrial pyruvate carrier dependent manner in TME. And the secretion of IL-10 promoted the proliferation and effector function of CAR-T cells, and achieved complete regression of established solid tumors and metastatic cancers in various solid tumor models (CRC, breast cancer, melanoma, pancreatic cancer). Moreover, IL-10 CAR-T cells triggered a stem cell like memory response in lymphoid organs, providing durable protection against tumor re-challenge [136].
In summary, cellular engineering strategies including CAR-T cell modification and targeted delivery, effectively address key challenges such as the immunosuppressive TME, impaired T cell homing, and T cell exhaustion. These approaches have demonstrated significant efficacy in treating IBD, CRC, and other related diseases, paving the way for tumor eradication and long term immune protection. Moving forward, they hold promise for enhancing cell based therapies, extending their application to broader diseases, enabling synergistic combinations with other treatments, ultimately improving therapeutic outcomes and patient survival.
4.3
Engineered fungus
Fungi have a wide range of applications in the treatment of colorectal related diseases because their natural metabolites have anti-inflammatory, antitumor, and immunomodulatory effects. And some of them are able to target intestinal foci, and regulate the balance of the bacterial flora [137,138]. However, natural fungi have shortcomings such as insufficient targeting, unstable metabolites and potential toxicity, which limit their clinical applications [139]. Some researchers are committed to the study of engineering modification. By using synthetic biology technology, it can optimize the metabolic pathway of fungi, improve the yield and stability of the active ingredients. At the same time, gene editing can give the fungi precise targeting ability, and reduce its toxicity. Since TME contains a large number of TAMs, and the mannose expressed in the natural yeast cell wall can target macrophages. Thus the yeast vector delivery system can achieve tumor targeted therapy by targeting TME [140].
An oral drug delivery strategy using yeast (Saccharomyces cerevisiae) loaded with alcohol dehydrogenase encapsulated ZIF NPs (ZIF@ADH) had been proposed. This yeast could target TAMs to accumulate in tumor tissues, and produce ethanol in a hypoxic environment, which was subsequently catalyzed by the enzyme to generate toxic acetaldehyde, which further promoted the production of inflammatory factors and M1 macrophage polarization to effectively inhibit CRC in situ. Besides, Zhang et al. proposed an innovative cancer therapeutic strategy of targeting in situ colorectal tumors by orally administering a Fungi based acetaldehyde generator (FBAG), which utilized the toxicity of acetaldehyde to induce apoptosis in cancer cells. The research team developed a fungal vector based on Saccharomyces cerevisiae (SC), which was genetically engineered to specifically metabolize ethanol to produce high concentrations of acetaldehyde in the hypoxic TME. The cytotoxicity of acetaldehyde was exploited to selectively kill tumor cells through local enrichment while avoiding systemic toxicity. The experimental results showed that after oral administration of the fungal vector, the engineered yeast could effectively colonize colorectal tumor sites, and continuously produce acetaldehyde in the presence of ethanol, significantly inhibiting tumor growth. In the animal model, the tumor volume of the treatment group was reduced by about 70%, and the survival of the mice was extended by >50%. And no significant liver or kidney function impairment or other systemic side effects were observed. Meanwhile, the therapy could further activate the anti-tumor immune response, and enhance the efficacy when combined with immune checkpoint inhibitors [141].
Meanwhile, yeast is an edible fungus, and the advantages of this oral drug device are its antitumor effect and good biosafety, which has great potential for clinical application. Zhang’s team proposed an innovative oral enzyme prodrug-immunotherapy combination for the treatment of CRC in situ. The team constructed a fungus triggered in situ chemotherapeutic drug generator called SC@CS@5-FC, which enabled targeted therapy via the oral prodrug 5-fluorocytosine (5-FC). When SC@CS@5-FC was targeted to the tumor via the tropism of SC, the chemotherapeutic generator could be catabolized by hyaluronidase (HAase), which was abundant in TME, to release the prodrug 5-FC. Subsequently, the non-toxic 5-FC was converted into the toxic chemotherapeutic drug 5-fluorouracil (5-FU) catalyzed by cytosine deaminase (CD) of SC. Meanwhile, SC and zinc with chitosan NPs acted as immune adjuvants to activate antigen presenting cells, further enhancing the therapeutic effect. The experimental results showed that SC@CS@5-FC could effectively inhibit tumor growth, and prolong the survival of mice [142].
With the rapid development of nanobiotechnology, some researchers have focused on combining fungi with nanomaterials to transport therapeutic nanomaterials to the lesion to maximize the effectiveness of the drugs. For example, Shen et al. developed an oral nano/gene delivery system based on engineered modified Saccharomyces cerevisiae for the treatment of CRC. The study modified Saccharomyces cerevisiae by genetic engineering techniques to display specific nanocarriers on its surface, which could effectively load and protect therapeutic genes (e.g., siRNA or plasmid DNA) for targeted delivery to colorectal tumor sites through the harsh environment of the gastrointestinal tract. The research team took advantage of the natural acid resistance and mucus penetrating ability of yeast to overcome the instability of conventional oral delivery systems in the presence of gastric acid and intestinal enzymes, while enhancing the specific recognition and enrichment of tumor tissues by surface modified targeting ligands, such as anti-EpCAM antibodies or folate receptor ligands. Experimental results showed that this delivery system exhibited excellent gene transfection efficiency in both in vitro and in vivo models, significantly silencing oncogenes (e.g., KRAS or β-catenin) or activating oncogenic pathways, thereby inhibiting tumor proliferation, and inducing apoptosis. In addition, the system demonstrated good biocompatibility and intestinal barrier penetration without causing significant immune response or toxicity [143]. This study provided an innovative strategy for the non-invasive treatment of CRC, combining the dual advantages of oral drug delivery and gene therapy precision, and layed an important foundation for clinical translation.
Engineered fungi hybrid systems enable precise treatment of colorectal related diseases through optimized production of bioactive metabolites and targeted delivery of drugs or genes, offering high therapeutic efficacy and biosafety. This approach opens new avenues for non-invasive treatment and clinical translation of colorectal related diseases, providing innovative strategies and technical support.
4.4
Engineered probiotics
Probiotics are a group of microorganisms that have significant health promoting effects on their hosts, and have received extensive attention in the healthcare field in recent years. Among them, Lactobacillus and Bifidobacterium, as the most representative probiotics, are widely distributed in natural environments such as fermented foods, human intestines and plants. Studies have shown that these probiotics play their physiological roles by regulating intestinal microecological balance, enhancing host immune function, promoting metabolic homeostasis, and other mechanisms, and also exhibit significant antimicrobial activity and antitumor potential [144,145]. Their mechanism of action mainly involves molecular pathways such as adhesion protein mediated colonization, bacteriocin secretion, and immunomodulation, which have important application prospects in cancer therapy and intestinal disease intervention. However, traditional probiotic intervention strategies, such as probiotic supplementation and fecal bacterial transplantation, still have some limitations in practical applications because the molecular mechanisms of host and microbiota interactions are not well understood.
In recent years, engineered in vivo therapeutic strategies that utilize probiotics to reconstitute delivery of drug molecules or mediate diagnostic methods have become a research frontier in the fields of biomedical engineering and synthetic biology [146]. Suppression of inflammation through oral administration of intestinal probiotic drugs is a common therapeutic strategy [147]. However, limitations such as poor gastric acid tolerance and low intestinal adhesion of probiotics limit their efficacy. In order to overcome the limitations of probiotic activity in acidic environments and short retention time in the intestine, Wang et al. developed a tumor specific acoustic sensitizer supply system based on engineered probiotic bacteria. The team selected the non-pathogenic E. coli strain Nissle (EcN), genetically engineered it with a recombinant plasmid, optimized the 5-aminolevulinic acid (5-ALA) biosynthesis pathway via synthetic biology, and then encapsulated the engineered EcN with 4T1 tumor cell membrane to construct a 5-ALA supply system based on engineered probiotics (SPECS). This system could synthesize 5-ALA efficiently, and was stable in a hypoxic TME. The tumor cell membrane encapsulation increased the uptake of tumor cells by SPECS. Combined with ultrasound, it significantly reduced the cell survival rate, and enhanced the generation of ROS. In vivo, tumor growth was inhibited by injecting 4T1 tumor bearing mice. And it induced immunogenic cell death (ICD), increased the number of M1 macrophages, DCs, and CD8+ T cells, and enhanced anti-tumor immunity [148].
Through the modified probiotics, they can precisely deliver therapeutic drugs to tumors, achieve non-invasive and efficient anti-cancer, and advance the development of precision cancer therapy. Recently, some studies have modified probiotics in combination with metallic nanomaterials for delivering anti-inflammatory molecules, and regulating the balance of intestinal flora. For example, Guo et al. found that the prevalence of Lactobacillus spp. in UC patients was reduced, and the level of oxidative stress was elevated [149], which correlated with the severity of inflammation, through machine learning and bioinformatics analysis. A probiotic based therapeutic approach was developed that could synergistically restore gut redox and microbiota homeostasis. A polysaccharide network was provided for the spatial crystallization of ultra-small, highly active selenium dots by inducing the formation of extracellular membranes in Lactobacillus casei (Lac). After oral administration of this engineered probiotic, the selenium dots embedded in the pericellular membrane enhanced the tolerance of Lac to gastric acid and intestinal mucus adhesion. At the lesion site, selenium dots effectively scavenged colonic ROS, and reduced the level of inflammatory factors. Lac regulated the intestinal flora, increased the abundance of beneficial bacteria (e.g., Lactobacillus spp.), and inhibited the proliferation of pathogenic bacteria. The synergistic effect attenuated the colonic mucosal injury and histopathological lesions. In various mouse models and non-human primate experiments, it significantly improved symptoms, and restored intestinal redox homeostasis and flora homeostasis. The dual-action mechanism of this therapy broken through the limitations of traditional probiotics with a single action, and the successful validation in non-human primates provided strong support for its clinical translation [150].
Meanwhile, in a study on the treatment of colitis, Li and other researchers developed Se@EcNnullC2/A2 by using clinically recognized EcN as a basis for intracellular synthesis of Se NPs and then encapsulating it, and developed the triple function probiotic. At the cellular level, the metabolites of Se@EcNnullC2/A2 were involved in the synthesis of related substances, scavenging of oxygen species, and inhibiting inflammatory signaling pathways, and also activated specific signaling pathways to promote the conversion of M1 to M2 type macrophages, and to regulate macrophage phenotype. In animal experiments, in mice with dextran sulfate sodium (DSS) induced colitis, the probiotic could effectively remove oxidative stress, inhibit inflammation, and repair the intestinal barrier. At the same time, EcN in the colon reduced Escherichia Shigella levels, and regulated the imbalance of the intestinal microbiota. Overall, Se@EcNnullC2/A2 demonstrated multiple favorable effects in the treatment of colitis, providing a new way for the treatment of colitis [151]. In addition, Hu’s team designed the engineered probiotic POSR@EcN by co-loading probiotic EcN with CO/H2S releasing copolymer (POSR) to achieve intestinal delivery of gas signaling molecules (Fig. S2 in Supporting information). POSR@EcN showed enhanced colonization ability in the gastrointestinal tract. Experiments in a mouse IBD model showed that after oral administration and supplemented with ultrasound treatment, CO and H2S were released locally at the site of inflammation, promoting the conversion of M1 to M2 type macrophages, inhibiting the expression of inflammatory mediators iNOS and TNF-α, and thus alleviating intestinal inflammation. In addition, CO/H2S delivery increased the expression of tight junction proteins ZO-1 and occludin, restored intestinal barrier integrity, and significantly increased the diversity of intestinal microbiota, promoting the growth of beneficial flora and the production of short chain fatty acids (SCFAs). Meanwhile, CO/H2S promoted the growth of Lactobacillus and Clostridium difficile, increased the levels of indoleacetic acid (IAA) and indole acetamide (IAM), regulated the gut-brain axis, inhibited microglia activation, alleviated anxiety- and depression-like behaviors, and restored cognitive functions in IBD mice [152].
In conclusion, engineered probiotics are capable of precisely targeting tumors or intestinal lesions, while exhibiting enhanced gastric acid tolerance and intestinal adhesion. They exert therapeutic effects by modulating the gut microbiota, exerting anti-inflammatory effects, and delivering therapeutic molecules, thereby minimizing systemic toxicity, and overcoming the limitations of conventional probiotics, such as their limited functionality, poor tolerance, and inadequate targeting. This approach represents a novel solution for the treatment of intestinal diseases, and provides new insights into the management of other microecology related disorders. It is expected to further advance research on microecological therapies, and accelerate their clinical translation.
4.5
Engineered phages
Natural phages are a class of viruses that specialize in infecting bacteria, and are widely found in natural environments such as soil, water bodies, and animal intestines. They consist of a protein shell and internal nucleic acids (DNA or RNA) that can specifically recognize and infect host bacteria, eventually leading to bacterial lysis and death [153]. However, natural phages have the disadvantages of narrow host range, susceptibility to immune clearance, insufficient targeting, and the possibility of triggering drug resistance in various diseases. Through gene editing to expand the host range, modifying surface proteins to enhance targeting, combining immunomodulatory molecules to improve anti-tumor effects, or loading drugs to achieve synergistic therapy, the engineered phages are obtained, showing great potentials in biomedical fields [154].
To ensure the genetic stability of these modified phages within the body, thereby preventing reversion mutations or unintended mutations, researchers engineer the phage through a combination of rational design and directed evolution, employing multiple genome editing techniques such as the enhanced mutant phage assisted evolution system (eMPAE) may be employed in conjunction with cytosine deaminases to achieve targeted base substitutions. The introduction of elements such as highly efficient terminators can then be utilized to completely prevent reversion mutations [155]. Concurrently, innovative utilisation of the bacteriophage’s own epigenetic mechanisms (such as DNA methylation) may further maintain genomic integrity [156]. Alternatively, by employing intelligent screening methods based on metagenomic data (such as Meta-SIFT), variants capable of stably maintaining infectivity under high selective pressure can be directly selected, ensuring their evolutionary advantage within complex in vivo environments [157].
In recent years, several researchers have investigated the role of engineered phages in colorectal related diseases. For example, Baker et al. utilized engineered phages to achieve protein production and release in the mammalian intestine. The team chose to target the virulent T4 phage of intestinal non-pathogenic E. coli. The T4 phage promoters were first screened to identify promoters that allowed efficient expression of heterologous proteins, and had little effect on the titer of the T4 phage. And then the screened phages were genetically engineered to carry and express specific proteins. In vitro experiments showed that the modified phage could efficiently express heterologous proteins such as green fluorescent protein (sfGFP). In a mouse model of colitis, engineered phage expressing serine protease inhibitors significantly reduced pro-inflammatory enzyme activities and alleviated colitis symptoms. It also reduced weight gain and inflammation in diet induced obese mouse models, with no significant safety concerns reported, demonstrating good efficacy and potential safety [158]. In addition, Zheng et al. reported a NP-phage assembly mediated targeted nanomedicine for colon cancer, aiming to improve the efficacy of antitumor chemotherapy by modulating the intestinal microbiota. The phage could specifically recognize and lyse Clostridium nucleatum, and reduce protective autophagy. And the covalently attached irinotecan (IRT) NPs induced apoptosis in cancer cells. The dextran shell of the NPs could provide energy support to enhance the fermentation of Clostridium butyricum, further enhancing the anti-tumor efficacy. Compared with single and combined chemotherapeutic drugs, antibiotics and phage-free nanomedicines, this treatment showed better anti-tumor effects in an in situ colon cancer model [159]. Other studies have shown that Clostridium nucleatum (Fusobacterium nucleatum) is strongly associated with CRC development. Therefore, therapeutic strategies targeting Fusobacterium nucleatum are expected to be a breakthrough for improving chemotherapy resistance [160]. In one study, the phage was azide-modified using a bioorthogonal reaction, and then covalently attached to dextran NPs loaded with the chemotherapeutic drug irinotecan. And this engineered phage could be specifically enriched in CRC tumors colonized with F. nucleatum positive CRC tumors, decreasing the abundance of Fusobacterium nucleatum, increasing the content of short chain fatty acids, and ameliorating the chemoresistance [161].
Intestinal infections are a common condition that causes a variety of gastrointestinal and parenteral diseases. Although the use of phages to kill pathogenic bacteria is accurate and effective, the oral administration of phages for the treatment of intestinal infections remains challenging. The harsh conditions associated with the gastrointestinal tract can destroy ingested phages, and most phages become inactive within minutes of exposure to gastric acid. Therefore, methods capable of delivering sufficient numbers of active phages via oral administration are essential for the treatment of intestinal infections. Lin et al. explored the effects of infecting bacteria with phages encapsulated in a polymer nanocoating. Using Salmonella typhimurium targeted phage N1 as a model, after optimizing the multiplicity of infection (MOI), polymers were used to construct nanocoating to prepare In-phages. Under different conditions, the titers of In-phages were stable, and could be amplified continuously, while the titers of Ex-phages decreased significantly. Under different MOI values, In-phages had a better ability to bind bacteria, lysed bacteria faster, and cleared biofilm better than Ex-phages. In mouse intestinal infection and related reactive arthritis model, it could effectively inhibit intestinal pathogens, reduce the inflammatory response, and alleviate the symptoms of intestinal infection and related arthritis, and had a good safety profile, with no obvious adverse effects. Compared with naturally released phages, In-phages showed superior performance in amplification, binding and lysis of bacteria, high bioavailability in vivo, and good efficacy and safety for the treatment of intestinal infections and related arthritis, which provides a new referrence for phage therapy [162].
Phages can be used as engineered vectors to infect intestinal commensal bacteria for in situ production, and release of therapeutic proteins in the intestinal tract, which opens up a new direction for colorectal related diseases treatment and biopharmaceutical drug discovery, and provides innovative strategies and methods.
4.6
Engineered exosomes
Exosomes are nanoscale vesicles secreted by cells, carrying bioactive molecules such as proteins, RNA, lipids, which are involved in intercellular communication and signaling. As natural NPs with diameters in the range of 30–150 nm, exosomes are promising carriers for drug delivery [163]. Engineering modification can enhance the targeting, stability, and therapeutic effects of exosomes, and make them play a greater role in the fields of disease diagnosis, drug delivery, and immunomodulation [164]. Exosomes modified and transformed by engineering technology can release internally encapsulated therapeutic drugs, nucleic acids and other substances into tumor cells, enhancing the bodys’ immune response while exerting targeted therapy [165].
In one study, Zhou et al. developed an engineered exosome (iRGD-Exo-siBRIX1) decorated with iRGD, and loaded with BRIX1 specific siRNAs. In vivo experiments showed that it significantly inhibited the growth of CRC, and enhanced the efficacy of 5-FU chemotherapy. This study demonstrated that the engineered exosome targeting BRIX1 could activate the nuclear kernel stress-ribosomal protein L5 (RPL5)/RPL11-p53 signaling pathway to inhibit tumor progression, realizing the dual anticancer effect, which provided a novel strategy for cancer treatment, and was more effective in combination with chemotherapy [166]. In addition, Ren’s team focused on the application of exosomes in drug delivery, and developed engineered DC derived exosomes (EmDEX@GA) with homing ability, overexpression of PD-1 for immune checkpoint blockade (ICB), and STING agonist to enhance anti-tumor immunity. In a mouse model of in situ breast cancer, local administration of EmDEX@GA could remodel the immunosuppressive microenvironment of tumor draining lymph nodes (TDLNs), stimulate potent anti-tumor immunity, and inhibit primary tumors as well as lymph node and distant metastasis. Compared with systemic ICB, it had a better efficacy in inhibiting distant metastasis. The team had used engineered exosomes to precisely regulate the immune microenvironment of TDLNs, providing new targets and delivery tools for tumor immunotherapy, and promoting the research and translation of “precisely regulating the tumor immune microenvironment”, which is expected to be expanded to the study of immune microenvironment regulation in colorectal related diseases (Fig. S3 in Supporting information) [167].
Overall, although exosomes are naturally excellent drug delivery and intercellular communication vehicles, engineering modifications are required to overcome their inherent limitations of insufficient targeting specificity and restricted therapeutic efficacy. This enables precise targeting of pathological sites, enhances drug accumulation and therapeutic outcomes, reduces off-target toxicity to normal tissues, and can further potentiate treatment effects by activating specific pathways or remodeling the immune microenvironment. Such engineered exosomes play a pivotal role in precision medicine, and will facilitate advancements in related research and clinical translation.
5.
Conclusions and outlook
In summary, engineered organisms exhibit many possibilities, ranging from the development of biotherapeutic agents based on natural microorganisms and living cells to the effective treatment of previously incurable cancers and other persistent human diseases. In this review, it provides a comprehensive summary of recent advances in the application of representative engineered organisms to colorectal related diseases. Based on natural organisms, various engineering modification strategies are applied according to the characteristics of colorectal related diseases for rational on demand design to address the key pain points in current disease treatment. The organisms can be used as carriers to deliver drugs and functional synthetic materials to appropriate lesion sites, as well as acting as bioactive therapeutic agents themselves to synergistically improve the efficiency of diseases treatment.
Engineered organisms holds immense potentials as a revolutionary therapeutic approach within the medical field. However, its progression towards clinical application faces multifaceted challenges stemming from technological, regulatory, and biosafety considerations. Although a large number of research cases in recent years have demonstrated that these engineered organism based systems have shown good therapeutic effects in oral drug delivery, systemic drug delivery, etc., with high therapeutic efficiencies and promising applications for the precision treatment of colorectal related diseases, there are still a number of challenges that need to be carefully considered and resolved in order to promote their future clinical applications. Firstly, the in vivo biosafety and potential risks of engineered organisms constitute a primary concern. As an emerging therapeutic platform, engineered organisms offer breakthrough efficacy, whilst presenting complex risks absent in conventional drugs. However, their biological activity and self replicating capacity may pose pathogenic risks. Th off-target editing activity in gene editing tools (such as CRISPR-Cas9) could induce non targeted genetic modifications, potentially activating oncogenes or inactivating tumour suppressor genes, thereby increasing tumourigenesis risk. And horizontal gene transfer poses public health threats, as exogenous genes may transfer via vectors like plasmids to human or environmental microbes, spreading drug resistance or generating novel pathogens. Besides, the lack of long term safety data introduces uncertainty risks for clinical translation. Furthermore, fabrication and quality represent critical considerations for clinical application. As living therapeutic products, engineered organisms involve lengthy and highly complex production processes, with active components exhibiting inherent biological heterogeneity. Scaling up from laboratory scale to commercial production compliant with GMP standards often leads to batch-to-batch quality variations. Finally, the unique properties of engineered organisms such as self replication and in vivo persistence challenge traditional regulatory paradigms based on chemical drugs and biological products. Although regulatory bodies are progressively adapting assessment pathways, issues persist including inconsistent standards, limited experience, and delayed guideline updates. For instance, systematic and actionable guidance remains lacking in areas such as non-clinical study design for living biological therapeutics, defining biological efficacy, and assessing risks associated with combination therapies. Such uncertainties significantly prolong development timelines, and increase compliance costs.
Although there are enormous obstacles that must be overcome before clinical applications, the great potential of these engineered organisms is clear. Therefore, we believe that innovative strategies of artificial intelligence (AI) driven and assisted design, combined with technologies such as CRISPR-Cas gene editing, can achieve breakthroughs in related technical fields, and address the key issues mentioned above [168-170]. To this end, it is necessary to study the mechanism of action of engineered organisms in greater depth, optimize the modification strategy, focus on leveraging AI for multimodal analysis of clinical data to achieve precise preoperative diagnosis and prognostic prediction, develop intelligent engineered organsims capable of sensing the TME and releasing drugs sequentially for in situ treatment, overcome delivery challenges for oral CRISPR-Cas systems by creating efficient, stable oral nanodelivery systems that traverse gastrointestinal barriers to target colonic lesions for enhanced efficacy, and design synthetic microbial communities that precisely regulate the gut microbiome through multi-strain synergistic effects to suppress pathogenic bacteria. The final goal is to make breakthroughs in solving the problems of reducing systemic toxicity and other adverse events, realizing large scale production, establishing the corresponding evaluation system, and improving the efficiency of disease treatment, so as to vigorously promote biotherapeutic agents based on engineered organisms to move forward to the clinical application, and to bring more effective treatment to the general public. The program will be more effective for the patients.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statement
Jiahao Huang: Investigation, Funding acquisition. Yongfeng Lan: Formal analysis, Data curation. Lifa Chen: Methodology. Xiaoxuan Guan: Data curation. Rong Liang: Software. Jiatong Liang: Validation. Ming Gao: Project administration. Jialiang Gan: Data curation. Yun Guo: Validation. Sen Zhang: Methodology.
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (No. 82160902), Joint Project on Regional High-Incidence Diseases Research of Guangxi Natural Science Foundation (No. 2024GXNSFAA010143), the China Postdoctoral Science Foundation (No. 2024MD753921), the Clinical Research Climbing Program-Youth Science and Technology Star Project of the First Affiliated Hospital of Guangxi Medical University (No. YYZS2023009), the First-class Discipline Innovation-driven Talent Program of Guangxi Medical University, the “Medical Excellence Award” Funded by the Creative Research Development Grant from the First Affiliated Hospital of Guangxi Medical University, and the “Outstanding Medical Talents Cultivation Plan” of the First Affiliated Hospital of Guangxi Medical University.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.112151.
Figure 1
The main therapeutic mechanism for colorectal related diseases: immunoactivation and suppression, apoptosis/proliferation regulation, angiogenesis inhibition/promotion, colorectal microenvironmental effect, gut microbiota regulation, and epigenetic regulation.
Figure 2
Three primary therapeutic strategies for colorectal related diseases, namely non-surgical treatment, surgical intervention, and integrated management. Non-surgical approaches primarily involve conservative interventions, including medication, nutritional support, and endoscopic therapies, suitable for conditions such as active IBD. Surgical treatments focus on direct removal of lesions, addressing emergencies or refractory conditions such as CRC and intestinal obstruction. Integrated treatments pay attention to multidisciplinary collaboration, employing a “neoadjuvant therapy before surgery + surgical intervention + adjuvant therapy after surgery” model to enhance outcomes in complex cases like advanced CRC.
Figure 3
The classification of engineered organisms, and their design strategies. The engineered organisms are mainly divided as engineered bacteria, cells, fungus, probiotics, phages and exosomes.

 DownLoad:
DownLoad:

 下载:
下载:




 下载:
下载:
