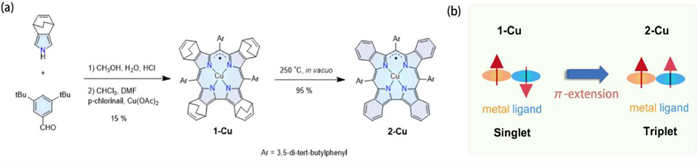
Figure 1.
(a) Synthesis route of 1-Cu and 2-Cu. (b) Schematic representation of the singlet-to-triplet conversion from 1-Cu to 2-Cu.

The coordination chemistry of corrole has attracted considerable interest from chemists in the past years [1]. Corrole is a ring-contracted porphyrin containing a directly linked C—C bond. With constricted central N4 cavities, corroles can act as closed-shell trianionic ligands to stabilize high-valence metal ions (MnⅤ, AgⅢ) [2,3], or exist as a dianionic radical ligands to balance the charge of coordinated ions (NiⅡ, ZnⅡ) [4,5]. In some cases, corrole show unique non-innocent character [6,7], and it is very difficult to achieve the accurate assignments of oxidation states of central metals, when coordinating to certain metals such as Cu, Co and Fe [8-11]. The unique electronic structures of non-innocent corrole complexes have offered them a variety of practical applications in the fields of spintronics [12,13], catalysis [14-17] and solar cells [18].
Among the reported non-innocent corrole complexes, the electronic structures of copper corroles have been one of the most controversial subjects. Copper corrole was initially considered to be a diamagnetic molecule containing a high-valent d8 Cu(Ⅲ) center [19] since it was both ESR-silent and NMR-active. However, the revisiting of the electronic structures of copper corroles by comprehensive spectroscopic and computational studies has suggested that their ground state is best described as an antiferromagnetically coupled d9 Cu(Ⅱ) ion and corrole radical [20,21]. Despite the Cu(Ⅱ)/Cu(Ⅲ) dilemma in copper corroles, Furuta group reported the unambiguous high-valent Cu(Ⅲ) ion on an N-confused corrole complex [22]. We have reported the detection of Kondo resonance of absorbed copper benzo-fused corrole [13], which was predicted to have a triplet ground state with ferromagnetically coupled d9 Cu(Ⅱ) ion and corrole radical. The ferromagnetic coupling configuration is in striking contrast with singlet states commonly identified in the regular copper corrole complex but has not been confirmed by common magnetometry measurements. In this work, we have synthesized a β-benzo-fused copper corrole 2-Cu having bulky substituents at meso-position using a retro Diels-Alder reaction (Fig. 1a) [23]. The comprehensive structural and magnetic data of 2-Cu, as well as theoretical calculations, establish a ground state conversion from singlet to an unusual highly stable triplet state via the extension of the corrole π-system (Fig. 1b).
A bicyclo[2.2.2]octadiene (BCOD) fused copper corrole 1-Cu was prepared through the condensation of the 4, 7-dihydro-4, 7-ethano-2H-isoindole and aldehyde catalyzed by hydrochloric acid in H2O/CH3OH mixture solvent [24]. The reaction mixture was extracted by CHCl3 subsequently, and the insertion of copper was achieved by adding copper acetate into the reaction mixture as a template during the oxidation process. Heating solid 1-Cu in vacuum gave corresponding β-benzo-fused corrole 2-Cu in 95% yield. The formation of 1-Cu and 2-Cu were confirmed by MALDI-TOF and ESI-HR mass spectra (Figs. S1–S4 in Supporting information). The introduction of bulky tert-butyl groups allows 2-Cu to have good solubility in common organic solvents (CH2Cl2, CHCl3, toluene, etc.).
The presence of paramagnetic species has a strong influence on NMR spectra, which can induce the line-boarding and additional paramagnetic chemical shifts of NMR peaks. 1-Cu exhibits well-resolved 1H NMR peaks in CD2Cl2, with aryl protons (Ha and Hb) in the region of 7.58–7.13 ppm and BCOD protons (Hc and Hd) in the region of 6.60–2.09 ppm, indicating the singlet character (Fig. 2a). By contrast, the aryl protons of 2-Cu appear in a broader range of 8.13–6.67 ppm, and the protons Hb show significant line broadening. Interestingly, we cannot find the 1H NMR signals of benzo protons in the expected chemical shifts range for diamagnetic molecules from +14 ppm to −4 ppm. Therefore, we extended the chemical shifts range of 1H NMR to −30 ppm, and eight broad peaks corresponding to benzo protons He were found in C6D6, which distributed from −6.2 ppm to −25.6 ppm (Fig. 2b). In the reported 1H NMR study of paramagnetic Fe(Ⅲ) corrole radicals, the signals of peripheral pyrrolic protons show a similar large upfield shift to the region ranging from −2 ppm to −60 ppm [25]. Using Evan's method [26,27], the magnetic susceptibility of the 2-Cu is determined to be 2.65 µB in CDCl3 solution at room temperature, confirming a high-spin S = 1 state (Fig. S7 in Supporting information).

Despite the radical nature, 2-Cu is highly stable and can be easily handled under ambient conditions, thus allowing for X-ray diffraction study. The single crystals suitable for X-ray diffraction measurements of 2-Cu were obtained from the slow diffusion of methanol into its CHCl3 solution. The top, side, and packing views of 2-Cu are shown in Fig. 3a. The C19N4 23-atom main corrole skeleton of 2-Cu takes a highly planar conformation with a mean plane deviation (MPD) value of only 0.024 Å. Based on the numerously reported X-ray diffraction analysis, the structure of copper corrole was considered to be inherently saddle-distorted due to the strongly antiferromagnetic coupling between Cu(Ⅱ) and corrole radical [28,29]. The planar structure of 2-Cu provides a perfect platform for ferromagnetic coupling of metal and ligand orthogonal orbitals [30]. The meso-phenyl groups of 2-Cu are nearly perpendicular to the main corrole plane with dihedral angles of 87.2°, 86.0° and 89.8°. Owing to the bulky tert-butyl groups, each 2-Cu molecule is substantially separated from the neighboring molecules with a large Cu-Cu distance of 11.2 Å, which facilitates its magnetic property investigation in solid form with negligible intermolecular interaction.

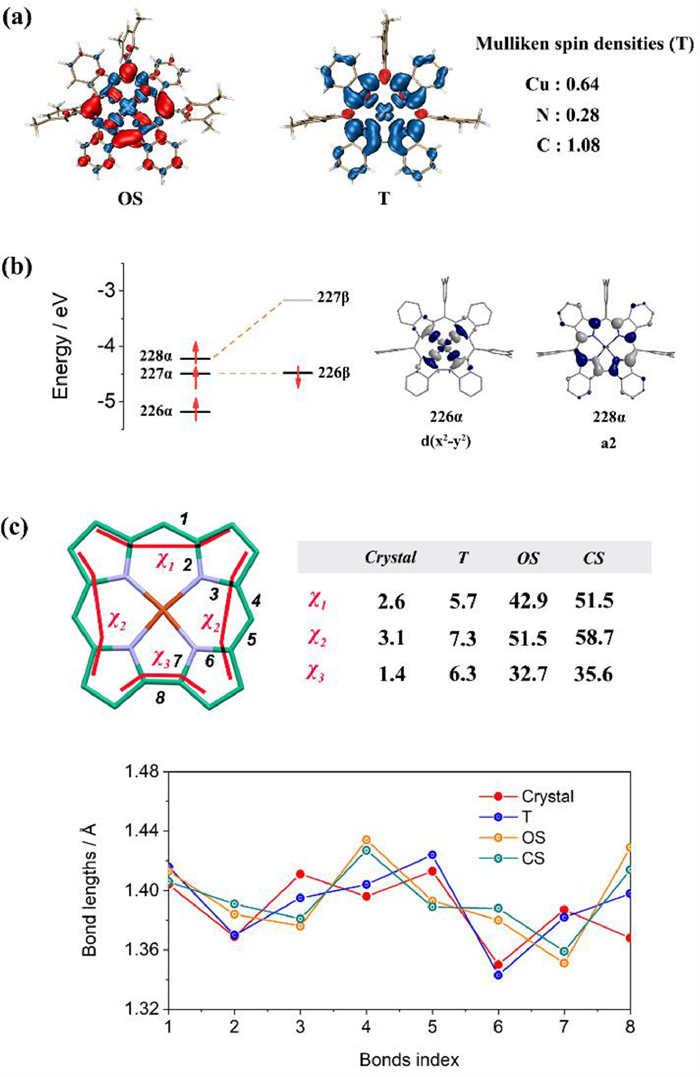
Density functional theory (DFT) calculations with B3LYP functional and 6–31G(d, p) bias set were performed on copper corroles for three different states: a close-shell Cu(Ⅲ) singlet (CS), an open-shell singlet antiferromagnetically coupled Cu(Ⅱ) corrole radical (OS) and a triplet ferromagnetically coupled Cu(Ⅱ) corrole radical (T) (Fig. 3b). The initial structure of 2-Cu for optimization was extracted from the single crystal diffraction data, with the tert-butyl groups replaced by methyl groups. The optimization was performed without symmetry constraint, and the resulting structures were analyzed by the vibrational frequency calculations to confirm the absence of any imaginary frequencies. The structures of two singlet states for 2-Cu are highly saddled, while the triplet state keeps the planar conformation. The triplet state was predicted to be the ground state of 2-Cu, bound with the open-shell singlet state 2.28 kcal/mol higher in energy, and the closed-shell state is 5.05 kcal/mol higher than the ground state. By contrast, the open-shell singlet state is predicted for the ground state of 1-Cu, which is consistent with the previously reported electronic structure of copper corrole [20,21]. The spin density maps of 2-Cu show that one spin distributes on d(x2-y2) orbital (blue), while another spin is distributed on the corrole b1 orbital (red) in the open-shell singlet state and on the corrole a2 orbital (blue) in the triplet state (Fig. 4a). The Mulliken spin populations of the triplet state of 2-Cu are 0.64 on Cu atom, 0.28 on N atoms, and 1.08 on C atoms, respectively. The frontier molecular orbitals of 2-Cu (Fig. 4b) show that the two unpaired electrons are delocalized on corrole a2 orbital (228α) and copper 3d(x2-y2) orbital (226α), respectively. The SOMO and SUMO are a pair of corrole a2 orbitals, indicating the ligand-based first oxidation and reduction. The torsion angles χ1, χ2 and χ3 of the crystal structure of 2-Cu are 2.6°, 3.1° and 1.4°, which in sharp contrast to the large estimated torsion angles of open-shell and closed-shell singlet states (42.9°/51.5°, 51.5°/58.7° and 32.7°/35.6°). The bond lengths from 1 to 8 are summarized, showing that the X-ray diffraction data exhibit better consistency with the theoretically predicted triplet structure than the singlet structures (Fig. 4c).
The paramagnetic property of 2-Cu was investigated for powder samples by temperature- and field-dependent superconducting quantum interference device (SQUID) magnetometry. The χT value of 2-Cu in 2 K is 0.77 cm3 K/mol, which is larger than the theoretical value of two uncoupled S = 1/2 spins (0.75 cm3 K/mol). No decrease in the χT values was observed at 2–300 K, suggesting the constant ferromagnetically coupling of the two spins. The observed χT–T plot was fitted with the Bleaney–Bowers singlet–triplet model [31-33] under a weak Weiss mean field (Weiss constant θ) (Fig. 5a). The singlet-triplet energy gap ΔEST was estimated to be +1.66 kcal/mol (J1/kB ≈ 424 K) with θ = −0.25 K, which is close to the theoretical value of ΔEST ≈ +2.28 kcal/mol. The field-dependent magnetization measurements at 2 K were fitted to a Brillouin function with S = 0.89, which is close to S = 1 and indicates its triplet ground state [32] (Fig. 5b). Interestingly, magnetic hysteresis of 2-Cu was observed at 2 K (Fig. 5c), suggesting its weak magnetic character [34]. We further investigated the magnetic properties 2-Cu by electron spin resonance (ESR) spectroscopy. The 2-Cu solutions with various solvents (toluene, THF and CH2Cl2) were all ESR-silent at both room temperature and 77 K, probably due to the large zero-field splitting. A similar ESR-silent phenomenon was also found on a ferromagnetically coupled Cu(Ⅱ) salen radical complex [35].

In summary, we have synthesized an air-stable copper tetrabenzocorrole radical 2-Cu bearing bulky groups at meso-position using a facile retro Diels-Alder reaction and identified its S = 1 triplet ground states both experimentally and theoretically. The ferromagnetically coupled Cu(Ⅱ)-corrole radical electronic configuration of 2-Cu was confirmed by NMR and SQUID data, as well as the distinct highly planar structure obtained from single-crystal X-ray diffraction. The DFT calculations predicted a low-lying triplet state for benzo-fused 2-Cu, and by contrast, an open-shell singlet ground state was precited for its precursor 1-Cu. Compared with the massively reported metallocorroles showing antiferromagnetically coupled singlet states, the example with triplet ground state is very rare. By benzo-fusion, the π-conjugation system of copper corrole was extended; hence the d-π interaction can be modulated to achieve a singlet-triplet conversion. This work provides a powerful tool for creating distinct and tunable spin systems for metallocorroles for the applications of novel magnetic and electronic devices.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by the National Natural Science Foundation of China (Nos. 22071103, 22001119 and 21911540069) and the Program A for Outstanding PhD Candidate of Nanjing University (No. 202201A006). The theoretical calculations are performed using the supercomputing resources at the High-Performance Computing Center of Nanjing University.
Supplementary material associated with this article can be found, in the online version, at
A. Ghosh, Chem. Rev. 117 (2017) 3798–3881. doi: 10.1021/acs.chemrev.6b00590
H. Shi, R. Liang, D.L. Phillips, et al., J. Am. Chem. Soc. 144 (2022) 7588–7593. doi: 10.1021/jacs.2c02506
C. Brückner, C.A. Barta, R.P. Briñas, et al., Inorg. Chem. 42 (2003) 1673–1680. doi: 10.1021/ic0261171
H. Gao, F. Wu, Y. Zhao, et al., J. Am. Chem. Soc. 144 (2022) 3458–3467. doi: 10.1021/jacs.1c11716
S. Will, J. Lex, E. Vogel, et al., Angew. Chem. 36 (1997) 357–361. doi: 10.1002/anie.199703571
Y.Y. Li, R.Z. Liao, Chin. Chem. Lett. 33 (2022) 358–361. doi: 10.1016/j.cclet.2021.06.028
S. Ganguly, A. Ghosh, Acc. Chem. Res. 52 (2019) 2003–2014. doi: 10.1021/acs.accounts.9b00115
K. Pierloot, H. Zhao, S. Vancoillie, Inorg. Chem. 49 (2010) 10316–10329. doi: 10.1021/ic100866z
S. Ganguly, D. Renz, L.J. Giles, et al., Inorg. Chem. 56 (2017) 14788–14800. doi: 10.1021/acs.inorgchem.7b01828
S. Ye, T. Tuttle, E. Bill, et al., Chem. Eur. J. 14 (2008) 10839–10851. doi: 10.1002/chem.200801265
S. Ganguly, L.J. Giles, K.E. Thomas, et al., Chem. Eur. J. 23 (2017) 15098–15106. doi: 10.1002/chem.201702621
J. Xu, L. Zhu, H. Gao, et al., Angew. Chem. 60 (2021) 11702–11706. doi: 10.1002/anie.202016674
F. Wu, J. Liu, P. Mishra, et al., Nat. Commun. 6 (2015) 7547. doi: 10.1038/ncomms8547
A. Rana, Y.M. Lee, X. Li, et al., ACS Catal. 11 (2021) 3073–3083. doi: 10.1021/acscatal.0c05003
H. Lei, H. Fang, Y. Han, et al., ACS Catal. 5 (2015) 5145–5153. doi: 10.1021/acscatal.5b00666
K. Sudhakar, A. Mahammed, Q.C. Chen, et al., ACS Appl. Energy Mater. 3 (2020) 2828–2836. doi: 10.1021/acsaem.9b02465
Q. Zhang, Y. Wang, Y. Wang, et al., Chin. Chem. Lett. 32 (2021) 3807–3810. doi: 10.1016/j.cclet.2021.04.048
A. Agresti, B. Berionni Berna, S. Pescetelli, et al., Adv. Funct. Mater. 30 (2020) 2003790. doi: 10.1002/adfm.202003790
I.H. Wasbotten, T. Wondimagegn, A. Ghosh, J. Am. Chem. Soc. 124 (2002) 8104–8116. doi: 10.1021/ja0113697
C.M. Lemon, M. Huynh, A.G. Maher, et al., Angew. Chem. 55 (2016) 2176–2180. doi: 10.1002/anie.201509099
M. Bröring, F. Brégier, E. Cónsul Tejero, et al., Angew. Chem. 46 (2007) 445–448. doi: 10.1002/anie.200603676
Y.K. Maurya, K. Noda, K. Yamasumi, et al., J. Am. Chem. Soc. 140 (2018) 6883–6892. doi: 10.1021/jacs.8b01876
S. Ito, T. Murashima, N. Ono, et al., Chem. Commun. (1998) 1661–1662.
B. Koszarna, D.T. Gryko, J. Org. Chem. 71 (2006) 3707–3717. doi: 10.1021/jo060007k
S. Cai, S. Licoccia, C. D'Ottavi, et al., Inorg. Chim. Acta 339 (2002) 171–178. doi: 10.1016/S0020-1693(02)00929-5
D.F. Evans, J. Chem. Soc. (1959) 2003–2005. doi: 10.1039/jr9590002003
F. Wu, J. Xu, H. Gao, et al., Chem. Commun. 57 (2021) 383–386. doi: 10.1039/d0cc06703b
A.B. Alemayehu, E. Gonzalez, L.K. Hansen, et al., Inorg. Chem. 48 (2009) 7794–7799. doi: 10.1021/ic900744v
K.E. Thomas, A.B. Alemayehu, J. Conradie, et al., Acc. Chem. Res. 45 (2012) 1203–1214. doi: 10.1021/ar200292d
H. Song, C.A. Reed, W.R. Scheidt, J. Am. Chem. Soc. 111 (1989) 6865–6866. doi: 10.1021/ja00199a070
B. Bleaney, K.D. Bowers, Proc. R. Soc. A 214 (1952) 451–465.
K. Kato, K. Furukawa, A. Osuka, Angew. Chem. 57 (2018) 9491–9494. doi: 10.1002/anie.201804644
K. Wang, P. Liu, F. Zhang, et al., Angew. Chem. 60 (2021) 7002–7006. doi: 10.1002/anie.202015356
D. Shimizu, A. Osuka, Angew. Chem. 57 (2018) 3733–3736. doi: 10.1002/anie.201801080
M. Orio, O. Jarjayes, H. Kanso, et al., Angew. Chem. 49 (2010) 4989–4992. doi: 10.1002/anie.201001040

Figure 1 (a) Synthesis route of 1-Cu and 2-Cu. (b) Schematic representation of the singlet-to-triplet conversion from 1-Cu to 2-Cu.

Figure 2 (a) 1H NMR spectra of 1-Cu and 2-Cu in CDCl3. (b) Selected range of negative values for the 1H NMR spectrum of 2-Cu in C6D6.

Figure 3 (a) Top, side and packing view of the single-crystal X-ray diffraction structure of 2-Cu. (b) DFT calculated structures and relative energies of 1-Cu and 2-Cu in triplet, open-shell singlet and closed-shell singlet states.

Figure 4 (a) Spin density distribution of 2-Cu. (b) Frontier molecular orbitals diagram of 2-Cu. (c) Selected bond lengths (Å) and dihedral angles (deg) of the crystal structure and optimized structures of 2-Cu in different spin states.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:

 下载:
下载: