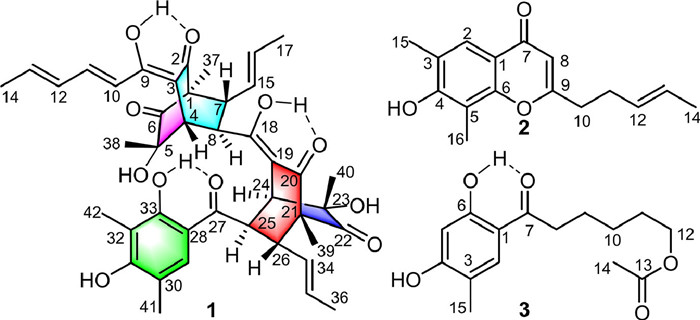
Figure 1.
Structures of compounds 1–3.
Citrinsorbicillin A, a novel homotrimeric sorbicillinoid isolated by LC-MS-guided with cytotoxic activity from the fungus Trichoderma citrinoviride HT-9
Guo-Ping Yin , Ya-Juan Li , Li Zhang , Ling-Gao Zeng , Xue-Mei Liu , Chang-Hua Hu

The sorbicillinoids are a family of hexaketide structures that have been reported to be a characteristic class of metabolites from fungal sources [1]. Structurely, these compounds typically have a highly malleable hexacyclic ring and a sorbyl side chain, which usually can be polymerized together through Michael addition or Diels-Alder reaction catalysed by enzymes, forming complex carbon skeletons with highly oxygenated and polycyclic system [2-5]. To date, approximately 177 natural sorbicillinoids have been reported, and they exhibit a variety of significant biological activities such as anticancer, radical-scavenging, and antimicrobial activities [6-14]. Amongst them, trimeric sorbicillinoids are very rare in nature and only six compounds have been reported from the deep-sea fungi Phialocephala and Penicillium [15-18], which have attracted extensive attention due to their unique structures and remarkable bioactivities [6-8].
In our ongoing endeavour to search for structurally unique and biologically interesting metabolites from fungal resources [19-21], we applied liquid chromatograph-mass spectrometer (LC-MS) analysis to screen endophytes from the traditional Chinese medicinal herb Coptis chinensis Franch. The LC/MS data of Trichoderma citrinoviride HT-9 revealed that this strain produced unusual sorbicillinoid metabolites, especially the minor ingredients with molecular weights approximately three times that of monomeric sorbicillinoids. Further high resolution electrospray ionization mass spectroscopy (HRESIMS)-guided dereplication analysis with subfractions found a unique molecule with a maximum ultraviolet (UV) absorption at 297 nm and a molecular ion [M + H]+ at m/z 729.3275 that did not match previously reported compounds, thus leading to obtain the first trimeric sorbicillinoid from terrestrial fungi, citrinsorbicillin A (1), along with two new monomeric sorbicillinoids citrinsorbicillins B (2) and C (3) (Fig. 1). 1 represented a unique carbon skeleton with two bicyclo[2.2.2]octanedione ring connected through an enolated carbon formed via two [4 + 2] cycloadditions. Moreover, 1 exhibited moderate inhibitory activity against human colon cancer HT29 cells. Herein, the details of their isolation, structural elucidation, and bioactivities, as well as plausible biogenetic pathways are reported.
Citrinsorbicillin A (1) was isolated as a yellow amorphous powder. The molecular formula was determined as C42H48O11 on the basis of a positive HRESIMS peak at m/z 729.3275 [M + H]+ (calcd. for C42H49O11, 729.3275), which required 19 degrees of unsaturation. The 1H nuclear magnetic resonance (NMR) data (Table 1) clearly exhibited 45 protons, including six singlet methyls (δH 0.92, 1.03, 1.11, 1.15, 2.11, 2.26), three doublet of doublets methyls [δH 1.52 (dd, 6.5, 1.8 Hz), 1.61 (dd, 6.5, 1.8 Hz), 1.95 (dd, 6.8, 1.8 Hz)], six aliphatic methines [δH 2.46 (d, 1.8 Hz), 2.77 (dd, 10.1, 7.4 Hz), 3.21 (dd, 7.4, 1.8 Hz), 3.32 (dd, 10.0, 7.0 Hz), 3.45 (d, 1.8 Hz), 4.36 (dd, 7.0, 1.8 Hz)], eight olefinic protons (δH 4.86–7.33), one aromatic singlet proton (δH 7.68), and three distinct hydroxyl groups (δH 13.17, 14.61, 14.04). The 13C NMR spectrum (Table 1) displayed a total of 42 resonances, which could be assigned with the aid of HSQC and DEPT data to nine methyls (δC 7.6, 9.8, 9.9, 16.0, 17.4, 17.8, 18.9, 23.4, 24.4), six sp3 methines (δC 41.1, 46.3, 46.8, 46.9, 47.1, 49.1), nine sp2 methines (δC 118.2, 127.9, 128.1, 129.9, 130.1, 130.6, 131.3, 139.4, 142.9), and 18 nonprotonated carbons (including nine olefinic carbons at δC 107.2, 107.3, 110.6, 112.2, 115.6, 159.5, 162.1, 169.6, 179.7; two oxygenated tertiary carbons at δC 75.0, 75.5; two quaternary carbons at δC 62.3, 62.9; and five keto groups at δC 197.0, 197.3, 203.2, 210.6, 211.1). Considering these NMR data and the molecular formula information with those of a known compound bisorbibutenolide isolated in this work [22], it was proposed that compound 1 was a sorbicillinoid homotrimer.
The planar structure of 1 was elucidated by comprehensive analysis of two-dimensional (2D) NMR data (Fig. 2). The key heteronuclear multiple bond connectivity (HMBC) correlations from H3–37 to C-1, C-2, C-6, and C-7, from H3–38 to C-4, C-5, and C-6, and from H-4 to C-2 and C-3, along with the 1H–1H correlated spectroscopy (COSY) correlations of H-4/H-8/H-7 constructed the first bicyclo[2.2.2]octanedione ring. Subsequently, the 1H–1H COSY correlations of H3–17/H-16/H-15/H-7 and H3–14/H-13/H-12/H-11/H-10, and the key HMBC correlations from OH-9 to C-3, C-9, and C-10 as well as the coupling constant of these related protons indicated that an (E)-isoallyl group was connected with the bicyclo[2.2.2]octanedione at C-7 and a (E, E)-penta-1, 3-diene group was further connected by a double bond between C-9 and C-3 to construct the fragment A. Similarly, the 1H–1H COSY correlations of H-24/H-25/H-26 and the key HMBC correlations from H3–39 to C-20, C-21, C-22, and C-26, from H3–40 to C-22, C-23, and C-24, and from H-24 to C-19 and C-20 constructed the second bicyclo[2.2.2]octanedione ring. The 1H–1H COSY correlations of H3–36/H-35/H-34/H-26 as well as the large coupling constant between H-34 and H-35 (15.0 Hz) further suggested that an (E)-isoallyl group was connected with the bicyclo[2.2.2]octanedione ring at C-26 forming the fragment B. Moreover, the fragment C was determined by the HMBC correlations from H3–41 to C-29, C-30, and C-31, from H3–42 to C-31, C-32, and C-33, from H-29 to C-27, C-28, and C-33, from OH-33 to C-28, C-32, and C-33. Finally, the key HMBC correlations from OH-18 to C-8, C-18, and C-19, from H-8 to C-18 and C-19, and from H-24 to C-18 and C-19 suggested that the fragment A fused to fragment B via the oxygenated olefinic carbon at C-18, while the fragments B and C were connected by a single bond between C-25 and C-27 based on the HMBC correlations from H-24 and H-26 to C-27 and from H-25 to C-27 and C-28. Hence, the planar structure of 1 was established.
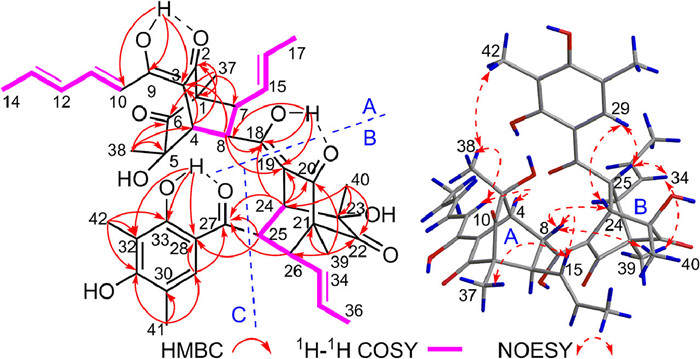
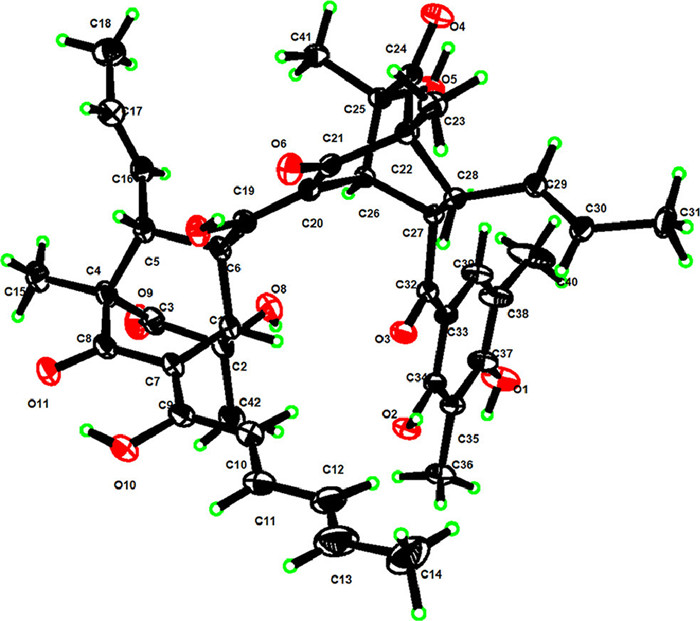
For the relative configuration of 1, the bicyclo[2.2.2]octanedione system (A) required the spatial orientation of the bridgehead-substituted H3–37 and H-4. The nuclear overhauser effect spectroscopy (NOESY) correlations (Fig. 2) of H3–38/H-10, H3–38/H3–42, H-10/H-4, and H-4/H3–38 suggested that these protons were proximal in space, while the correlations of H3–37/H-15 and H-15/H-8 indicated they were co-orientated. The large coupling constant of 3 JH-7/H-8 (7.4 Hz) further indicated the trans configuration between H-7 and H-8. Moreover, the small coupling constant (1.8 Hz) between H-4 and H-8 was similar to those of other sorbicillinoid analogues [6], thus defining their spatial position relationship. Similarly, the relative configuration of the bicyclo[2.2.2]octanedione system (B) was defined by analysis of the NOESY spectrum and coupling constant as shown in Fig. 2. Finally, the single-crystal X-ray diffraction (Fig. 3) analysis (Cu Kα) with the Flack parameter of 0.08(5) further confirmed the above deduction and determined the absolute configuration of 1 as 1R, 4S, 5S, 7R, 8R, 21R, 23S, 24S, 25R and 26R.
Citrinsorbicillin B (2) was obtained as a brown amorphous powder. The molecule formula C16H18O3 was established by HRESIMS at m/z 281.1154 [M + Na]+ (calcd. for C16H19O3, 281.1154), corresponding to eight degrees of unsaturation. The 1H NMR data (Table 1) of 1 showed signals for two singlet methyls (δH 2.33, 2.35), one doublets methyl [δH 1.64 (d, 5.5 Hz)], three olefinic protons (δH 5.46–6.11), one aromatic proton δH 7.83 (s), and four aliphatic protons (δH 2.43–2.69). The 13C NMR and HSQC data displayed 16 carbon resonances (Table 1), accounting for three methyls (δC 8.2, 15.9, 17.9), two sp3 methylenes (δC 29.8, 34.4), four sp2 methines (δC 109.4, 124.0, 126.9, 128.7), and seven nonprotonated carbons (110.7, 117.1, 121.8, 154.7, 156.7, 168.4, 178.5). In the 2D NMR spectra (Fig. 4), the 1H–1H COSY correlations of H3–14/H-13/H-12/H2–11/H2–10 and the key HMBC correlations from H2–10 to C-9 and C-8 and from H-8 to C-9 and C-10, as well as the large coupling constant between H-12 and H-13 (15.4 Hz) constructed an oxidized (E)-hepta-1, 5-diene moiety. The trisubstituted m-xylene moiety was confirmed by the key HMBC correlations from H3–15 to C-2, C-3, and C-4, from H3–16 to C-4, C-5, and C-6, and from H-2 to C-1 and C-6. Moreover, the HMBC correlations from H-2 to C-7 and from H-8 to C-1 and C-7 indicated that the (E)-hepta-1, 5-diene moiety at C-8 was connected to the trisubstituted m-xylene moiety at C-1 by a keto group at C-7. Considering the remaining unsaturation and the chemical shifts of C-6 (δC 154.7) and C-9 (δC 168.4), the oxo bridging between C-6 and C-9 was deduced to establish the core skeleton of chromogen. Thus, the structure of 2 was constructed.
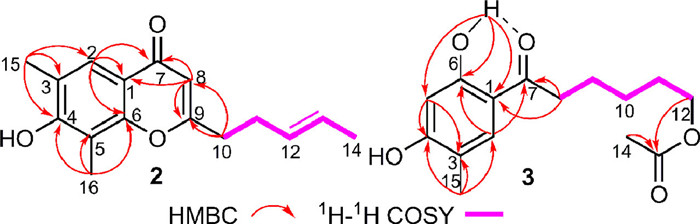
Citrinsorbicillin C (3) was obtained as a brown oil. Its molecular formula was deduced to be C15H20O5 with six degrees of unsaturation based on the protonated HRESIMS ion at m/z 303.1206 [M + Na]+ (calcd. for C15H20O5Na, 303.1208). The 1H NMR data (Table 1) of 3 showed signals for two singlet methyls (δH 2.05, 2.19), two typical triplet methylenes (δH 2.91, 4.09), two aromatic protons (δH 6.35, 7.47), and a hydroxyl group (δH 12.65). The 13C NMR and HSQC data displayed 15 carbon resonances (Table 1), accounting for two methyls (δC 15.1, 21.0), five sp3 methylenes (δC 24.3, 25.7, 28.5, 37.7, 64.4), two sp2 methines (δC 103.1, 132.2), and six nonprotonated carbons (113.5, 116.0, 161.1, 163.5, 171.4, 204.5). Comparison of its NMR data with those of 2 revealed they shared similar structural features. The major difference was that the pyranone ring was opened and reduced in 3, and the side chain was changed into an acetylated aliphatic chain, which were confirmed by the 1H–1H COSY correlations (Fig. 4) of H2–8/H2–9/H2–10/H2–11/H2–12 and the key HMBC correlations from H3–14 to C-13, from H2–12 to C-13, from H2–8 to C-1 and C-7, from H3–15 to C-2, C-3, and C-4, from OH-6 to C-1, C-5, and C-6, from H-5 to C-4 and C-6, and from H-2 to C-1 and C-7. Thus, the structure of 3 was elucidated.
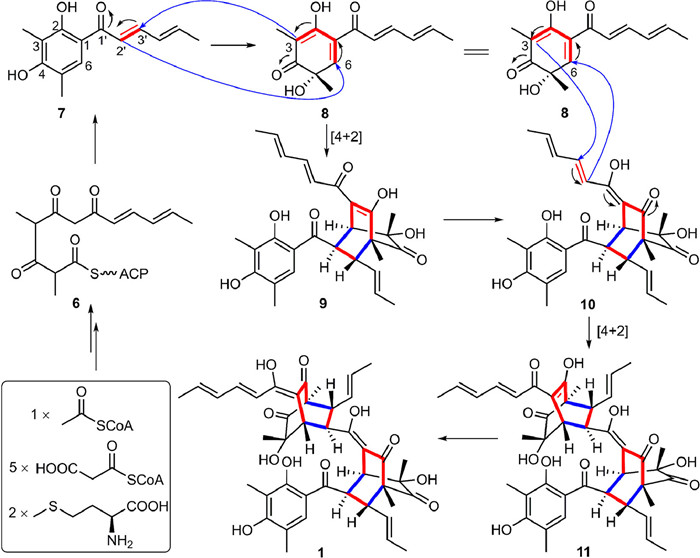
Citrinsorbicillin A (1) represents an unprecedented carbon skeleton, whose plausible biosynthetic pathway is postulated from three molecules of sorbicillion (7) by two [4 + 2] cycloadditions (Scheme 1) [6-8]. Specifically, the known precursors 7 and sorbicillinol (8) began to be biosynthesized through a polyketide biosynthetic pathway with an oxidation process [2]. Then, the first [4 + 2] cycloaddition between the sorbyl side chain in 7 at C-2′/C-3′ and the hexacyclic rings in 8 at C-3/C-6 constructed the intermediate 9, which subsequently converted to 10 through intramolecular keto-enol isomerization. Moreover, 10 coupled 8 to construct 11 via the second [4 + 2] cycloaddition, which further performed the similar keto-enol isomerization to form the backbone 1.
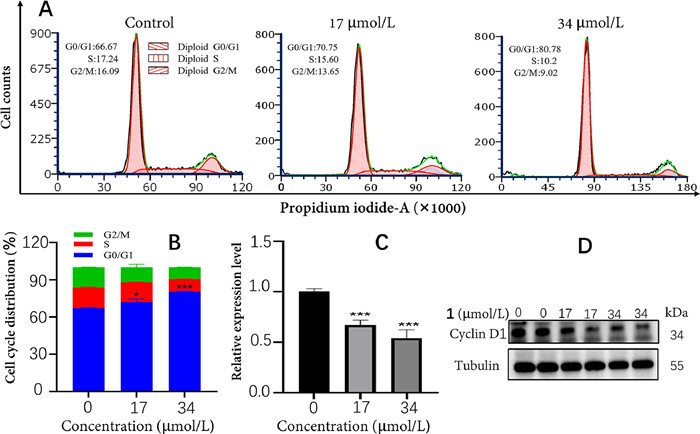
All of the isolates were evaluated for their cytotoxic activities against three human tumor cell lines (A549, HeLa and HT29) using methyl thiazolyl tetrazolium (MTT) method [23,24]. As a result (Table S1 in Supporting information), 1 exhibited moderate cytotoxicity against HT29 cells with half maximal inhibitory concentration (IC50) value of 34.0 µmol/L, compared with the positive control drug (oxaliplatin, IC50 = 45.5 µmol/L). To further explore whether 1 induced growth inhibition was associated with cell cycle regulation, PI staining analysis and cell cycle distribution were assessed by flow cytometry in HT29 cells. Compared to the control group, the percentage of G0/G1 phase cells in 1 was obviously increased at the concentrations of 17 and 34 µmol/L (Figs. 5A and B). The result indicated that 1 could induce cell cycle arrest at G0/G1 phase in a dose-dependant manner. Moreover, western blot analyses showed that the level of cyclin D1 protein significantly decreased after treatment with 1 (Figs. 5C and D). These data suggested that the cytotoxicity of 1 could be mediated by inhibiting the expression of cyclin D1 to block the cell cycle.
In summary, a novel homotrimeric sorbicillinoid citrinsorbicillin A (1) and two new related monomers 2 and 3 were isolated from the Coptis chinensis endophyte Trichoderma citrinoviride HT-9. 1 possessed an unprecedented carbon skeleton and showed moderate cytotoxicity against human colon cancer HT29 cells. These findings provided a novel molecular template for chemists and pharmacologists to further design and discovery of new anticancer agents.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by the National Natural Science Foundation of China (No. 82003629), the Natural Science Foundation of Chongqing (No. cstc2021jcyj-msxmX0975), and the Open Project of State Key Laboratory of Natural Medicines (No. SKLNMKF202304).
Supplementary material associated with this article can be found, in the online version, at doi:
D. Lai, J. Meng, X. Zhang, et al., Org. Lett. 21 (2019) 1311–1314. doi: 10.1021/acs.orglett.8b04101
L. Kahlert, E.F. Bassiony, R.J. Cox, E.J. Skellam, Angew. Chem. Int. Ed. 59 (2020) 5816–5822. doi: 10.1002/anie.201915486
T.M. Milzarek, M. Einsiedler, H. Aldemir, et al., Org. Lett. 21 (2019) 4520–4524. doi: 10.1021/acs.orglett.9b01398
A. Sib, T.A.M. Gulder, Angew. Chem. Int. Ed. 57 (2018) 14650–14653. doi: 10.1002/anie.201802176
A. Sib, T.A.M. Gulder, Angew. Chem. Int. Ed. 56 (2017) 12888–12891. doi: 10.1002/anie.201705976
A.M. Harned, K.A. Volp, Nat. Prod. Rep. 28 (2011) 1790–1810. doi: 10.1039/c1np00039j
J. Meng, X. Wang, D. Xu, et al., Molecules 21 (2016) 715. doi: 10.3390/molecules21060715
X. Hou, X. Zhang, M. Xue, et al., J. Fungi 8 (2022) 62. doi: 10.3390/jof8010062
X.Y. Guo, H.T. Li, Y.T. Shao, et al., Fitoterapia 166 (2023) 105443. doi: 10.1016/j.fitote.2023.105443
K.A.U. Zaman, X. Wu, A.M. Sarotti, S. Cao, Nat. Prod. Res. 36 (2022) 5984–5990. doi: 10.1080/14786419.2022.2056890
X.P. Peng, G. Li, L.M. Wang, et al., Front. Microbiol. 13 (2022) 800626. doi: 10.3389/fmicb.2022.800626
X. Pang, P. Wang, S. Liao, et al., Phytochemistry 202 (2022) 113311. doi: 10.1016/j.phytochem.2022.113311
S.Z. Liu, G.X. Xu, F.M. He, et al., Mar. Drugs 20 (2022) 213. doi: 10.3390/md20030213
C. Duan, S. Wang, R. Huo, et al., J. Fungi 8 (2022) 530. doi: 10.3390/jof8050530
M.J. Cao, T. Zhu, J.T. Liu, et al., Nat. Prod. Res. 34 (2020) 2880–2886. doi: 10.1080/14786419.2019.1596099
W. Guo, J. Peng, T. Zhu, et al., J. Nat. Prod. 76 (2013) 2106–2112. doi: 10.1021/np4006647
D. Li, S. Cai, T. Zhu, et al., Tetrahedron 66 (2010) 5101–5106. doi: 10.1016/j.tet.2010.04.111
D. Li, F. Wang, X. Xiao, et al., Tetrahedron Lett. 48 (2007) 5235–5238. doi: 10.1016/j.tetlet.2007.05.134
G.P. Yin, M. Gong, G.M. Xue, et al., J. Nat. Prod. 84 (2021) 2623–2629. doi: 10.1021/acs.jnatprod.1c00185
G.P. Yin, Y.R. Wu, C. Han, et al., Org. Chem. Front. 5 (2018) 2432–2436. doi: 10.1039/c8qo00070k
G.P. Yin, Y.R. Wu, M.H. Yang, et al., Org. Lett. 19 (2017) 4058–4061. doi: 10.1021/acs.orglett.7b01823
N. Abe, T. Murata, A. Hirota, Biosci. Biotechnol. Biochem. 62 (1998) 2120–2126. doi: 10.1271/bbb.62.2120
S. Lin, J. Huang, H. Zeng, et al., Chin. Chem. Lett. 33 (2022) 4587–4594. doi: 10.1016/j.cclet.2022.03.064
Y. Peng, Y. Chang, C. Sun, et al., Chin. Chem. Lett. 33 (2022) 4261–4263. doi: 10.1016/j.cclet.2022.03.003

Figure 5 The cytotoxicity analyses of 1 in HT29 cells. (A) Flow cytometry analysis of HT29 cells treated with 17 and 34 µmol/L of 1 for 48 h. (B) Percentage of cell cycle distribution of HT29 cells treated with different concentrations of 1. (C) The relative expression level of cyclin D1 in HT29 cells treated with 17 and 34 µmol/L of 1. (D) Western blot analysis of cyclin D1 protein expression of 1. The data are presented as the means ± SEM of three replicates. P < 0.05, ***P < 0.001, compared with the control group.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:


 下载:
下载:




 下载:
下载: