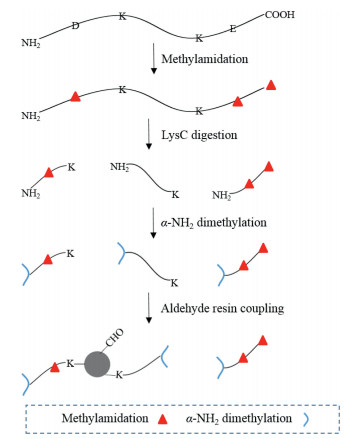
Scheme 1.
Workflow of MADMAR for identification of C-terminal peptides.

The carboxyl terminus, which is also abbreviated as C-terminus, is the end of every protein. The C-termini of proteins are often disordered and solvent exposed [1]. In addition, proteins may be modified through proteolysis in vivo, thus generating new C-termini [2], which further increases the diversity of C-terminome. These properties enable C-termini to participate in different biological processes. For instance, the C-terminal tail of receptor plays an important role in high-affinity arrestin binding [3]. Also, the conformation of C-terminus of ferroportin changes at different Mn2+ concentrations [4]. In addition, RNA splicing, post-translational modification, and proteolytic processing of proteins in vivo could lead to protein neo-C-termini [5]. Despite the known important roles of C-termini, numerous biological functions of C-termini still remain undiscovered. Thus, in-depth C-terminome analysis technologies are critical for understanding the nature of C-termini-associated events.
Mass spectrometry (MS) has been widely used for analysis on peptide level [6]. However, highly selective analysis of C-teminome based on MS is still impeded by the inherent properties of C-termini. For example, the ionization of C-terminal peptides during mass spectrometric analysis is often not efficient because of their negative charges and lack of basic residues [7]. Various attempts have been made to address the problem. Chen et al. labeled the carboxyl groups with methylamine to neutralize their negative charges [8]. Kaleja et al. increased the charges of C-termini to boost their identification by introducing the positive charge molecules of N, N-dimethylethylenediamine (DMEDA) [9]. In addition, C-terminal peptides only comprise a small proportion of the digested peptides. Therefore, enriching the C-terminal peptides is also necessary. There are two main ways of enriching C-terminal peptides, namely positive enrichment and negative enrichment. Positive enrichment involves enriching C-terminal peptides on the functionalized material, while leaving the other peptides in the solution. Unfortunately, low reactivity of the carboxyl group and lack of site-specific labels of carboxyl group on the C-terminus hamper the development of positive enrichment strategies [10]. Therefore, the prevalent enrichment strategy is based on negative enrichment.
Negative enrichment strategies often used functionalized material that can remove the undesired peptides, while leaving C-terminal peptides in the solution. A variety of resin-based negative enrichment strategies have been proposed. In a general negative enrichment workflow, for example the C-TAILS [11], the amine and carboxyl groups of proteins were protected first. Then proteins were subjected to enzymatic digestion to generate peptides mixture. The digests were then coupled with amine-functionalized resin to capture all internal peptides containing free carboxyl group, while leaving the blocked protein C-terminal peptides in the supernatant. Finally, the supernatant was collected and can be fractionated to reduce the sample complexity for MS analysis. In order to identify more protein C-termini, Zhang et al. systematically optimized the conventional C-TAILS method, which yields 57% more C-termini than the original one using E. coli sample [12]. Hu et al. chose LysargiNase as the enzyme to provide an extra positive charge on the C-terminal peptides compared with C-TAILS, thus facilitating the identification of C-terminal peptides. As a result, they identified a total of 834 C-termini from three fractions of proteome of 293T cell [13]. Du et al. combined LysC digestion and site-selective dimethylation to remove N-terminal and internal peptides by aldehyde resin, and a total of 781 C-termini from HeLa cell were identified from six fractions [14].
Herein we report an alternative negative enriching method that incorporates methylamidation, site-selective dimethylation and aldehyde resin coupling (MADMAR) for in-depth C-terminome analysis (Scheme 1). In our method, protein disulfide bonds are reduced and alkylated. Then, all the carboxyl groups on the protein are amidated with methylamine, followed by LysC digestion of methylamidated proteins. After that, site-selective dimethylation on the N-terminal amino groups of digested peptides are performed without affecting the side chain amino groups of lysine (Schematic diagram see Scheme S1 in Supporting information) [15]. Finally, aldehyde resin coupling reaction is performed to remove peptides other than C-terminal peptides. The C-terminal peptides in the supernatant are subjected to mass spectrometry analysis. By using the new workflow, we achieved in-depth analysis of the C-terminome from HeLa cell without fractionation. Importantly, we can obtain the database-annotated C-termini and neo-C-termini simultaneously in this workflow, whereas the neo-C-termini cannot be obtained in some previously developed workflow including ours [14].
Methylamidation can label all carboxyl groups regardless of their position. To check the methylamidation reaction efficiency, we used a synthetic peptide with the sequence of TPVEPEVAIHR ([M+H]+ m/z 1247.7) as a model. After methylamidation reaction, the original peptide peak disappeared while another peak emerged at m/z 1286.8, indicating all of the three carboxyl groups were completely methylamidated (Figs. S1a and b in Supporting information). The MS/MS spectrum further verified that the new peak can be attributed to fully methylamidated peptide (Fig. S2 in Supporting information).
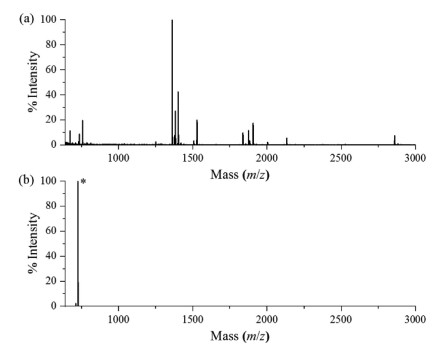
The LysC digestion condition, site-selective dimethylation reaction and aldehyde resin coupling reaction have been systematically optimized in our previous work [14]. Therefore, after evaluation of the methylamidation reaction, we directly incorporated them into the MADMAR method and evaluated the newly developed method using a standard protein myoglobin as a model. In brief, 50 µg myoglobin was methylamidated and digested with LysC. Then the digested peptides were site-selectively dimethylated, followed by aldehyde resin coupling. As shown in Fig. 1a, without MADMAR treatment, the peak of C-terminal peptide from myoglobin (ELGFQG, [M+H]+ m/z 650.3) was almost completely obscured by other peptides. After enrichment, the C-terminal peptide was detected as [M+Na]+ at m/z 726.5, which dominated in the spectrum (Fig. 1b). The mass spectrum shows that peptides other than myoglobin C-terminal peptide only constituted a small proportion of the enriched peptides. The MS/MS spectrum verified that the peak (m/z 726.5) can be attributed to methylamidated and dimethylated myoglobin C-terminal peptide (Fig. S3 in Supporting information).
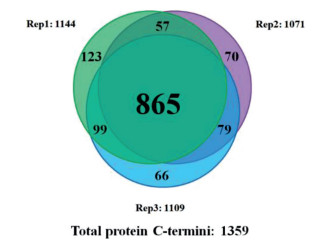
We first checked the selectivity of site-selective dimethylation on HeLa proteome, 85.2% of the identified peptides were desired products. Then we used MADMAR method to analyze the C-terminome of human HeLa cells. As a result, we identified 1638 protein C-termini from technical triplicates (Tables S1 and S2 in Supporting information), 1359 of them were database-annotated protein C-termini. Eighty-one percent of the database-annotated protein C-termini were identified in at least two replicates. (Fig. 2) Ninety point five percent of database-annotated C-terminal peptides were methylamidated at all carboxyl groups.
To compare with our previous work based on aldehyde resin [14], 79% of the database-annotated protein C-termini identified in our previous work were also identified in MADMAR, exhibiting good coverage of MADMAR. Meanwhile, although fractionation was eliminated in MADMAR, MADMAR still yielded 74% more database-annotated protein C-termini from HeLa cell with lower protein amount (50 µg) than our previous work (300 µg) (Fig. S4 in Supporting information). We also analyzed the distribution of the histidine numbers in the identified annotated protein C-termini. No significant bias was observed in terms of histidine-containing numbers (Fig. S5 in Supporting information). These results demonstrated that MADMAR could be an effective and unbiased method for biological samples C-terminome analysis.
In addition, with the aid of methylamine label on protein C-termini, MADMAR could identify protein neo-C-termini, thus 279 neo-C-termini were identified from HeLa cells. Some of them have various neo-C-termini forms. For instance, Glyceraldehyde-3-phosphate dehydrogenase (P04406) has eight different C-terminal forms. Prosaposin (P07602) has seven different C-terminal forms. To discover the possible cleavage events, the neo-C-termini were analyzed using IceLogo [16]. As shown in Fig. 3, Cys was predominant at the position next to neo-C-termini, while Arg, Asn, Glu and Gln were often the first amino acid at the neo-C-termini. The overrepresentation of Asn at the position 0 was also reported in previous observations [13, 17].

In summary, we developed a method that incorporated MADMAR for in-depth profiling the C-terminome. We identified 1638 protein C-termini from 50 µg HeLa cell proteins using this method, of them 279 were protein neo-C-termini. Our method has these advantages: (ⅰ) Methylamidation on the protein level enables identification of database-annotated C-termini and neo-C-termini simultaneously. (ⅱ) Through removing the internal peptides, extensive fractionation of the targeted C-terminome is eliminated, which greatly reduced the total analysis time while achieved better identification with lower sample amount. This method may provide a new tool for protein C-terminome analysis and pave the way for discovering more biological functions of protein C-termini.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
The work was supported by the National Key Research and Development Program of China (No. 2017YFA0505001), National Natural Science Foundation of China (No. 21974025) and the project of Shanghai Key Laboratory of Kidney and Blood Purification.
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.08.022.
S. Sharma, M.R. Schiller, Crit. Rev. Biochem. Mol. Biol. 54(2019) 85-102. doi: 10.1080/10409238.2019.1586828
C. Lopez-Otin, C.M. Overall, Nat. Rev. Mol. Cell. Biol. 3(2002) 509-519. doi: 10.1038/nrm858
X.E. Zhou, Y. He, P.W. de Waal, Cell 170(2017) 457-469. doi: 10.1016/j.cell.2017.07.002
M.G. Zhang, M. Wen, Y. Xiong, L.H. Zhang, C.L. Tian, Chin. Chem. Lett. 29(2018) 1509-1512. doi: 10.1016/j.cclet.2018.04.026
N. Fortelny, S.R. Yang, P. Pavlidis, P.F. Lange, C.M. Overall, Nucleic Acids Res 43(2015) D290-D297. doi: 10.1093/nar/gku1012
S.X. Cao, J.C. Zhang, M.Y. Niu, Chin. Chem. Lett. 15(2004) 652-654.
P.F. Huesgen, P.F. Lange, L.D. Rogers, Nat. Methods 12(2015) 55-58. doi: 10.1038/nmeth.3177
L. Chen, Y. Shan, Y. Weng, Anal. Bioanal. Chem. 408(2016) 3867-3874. doi: 10.1007/s00216-016-9476-1
P. Kaleja, A.O. Helbig, A. Tholey, J. Proteome Res. 18(2019) 2954-2964. doi: 10.1021/acs.jproteome.9b00264
M. Liu, C. Fang, X. Pan, Anal. Chem. 87(2015) 9916-9922. doi: 10.1021/acs.analchem.5b02437
O. Schilling, O. Barre, P.F. Huesgen, C.M. Overall, Nat. Methods 7(2010) 508-511. doi: 10.1038/nmeth.1467
Y. Zhang, Q. He, J. Ye, Anal. Chem. 87(2015) 10354-10361. doi: 10.1021/acs.analchem.5b02451
H. Hu, W. Zhao, M. Zhu, Anal. Chem. 91(2019) 14522-14529. doi: 10.1021/acs.analchem.9b03543
X. Du, C. Fang, L. Yang, Anal. Chem. 91(2019) 6498-6506. doi: 10.1021/acs.analchem.8b05338
H. Qin, F. Wang, Y. Zhang, Chem. Commun. 48(2012) 6265-6267. doi: 10.1039/c2cc31705b
N. Colaert, K. Helsens, L. Martens, J. Vandekerckhove, K. Gevaert, Nat. Methods 6(2009) 786-787. doi: 10.1038/nmeth1109-786
Q. Li, Y. Zhang, J. Huang, Anal. Chem. 92(2020) 4742-4748. doi: 10.1021/acs.analchem.9b05280

Figure 1 MALDI-TOF mass spectra of myoglobin digested by LysC. (a) Direct analysis. (b) After MADMAR treatment. The C-terminal peptide peak was marked with *.

Figure 2 Overlap of database-annotated protein C-termini in technical triplicates of MADMAR on HeLa proteome.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:

 下载:
下载: