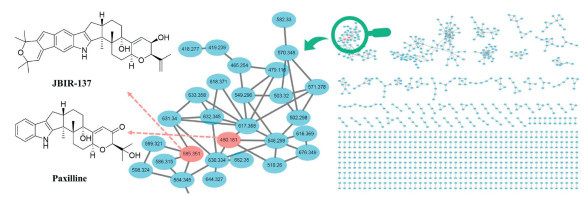
Figure 1.
LC-MS-based molecular networking of the EtOAc extract of fungus P. janthinellum H-6.
Penispirolactam and penipyrroloindole, two unusual polycyclic indole diterpenoids with anti-hepatic fibrosis activity from the endophytic fungus Penicillium janthinellum H-6
Lei Li , Xin-Ying Zhu , Jun-Yu Zhu , Zhi-Hao Wu , Yan-Jiang Zhang , Fang-Yu Yuan , Dong Huang , Sheng Yin , Gui-Hua Tang

Natural products with distinctive structural frameworks have long served as a cornerstone of groundbreaking drug discovery [1,2]. Indole diterpenoids (IDTs) from fungi represent a growing class of structurally complex special natural products with remarkable biological properties [3-7], including insecticidal, antibacterial, antiviral, cholesterol-lowering, and blood glucose-regulating activities. Among numerous IDTs, the paspaline-type IDTs are characteristic of a unique six ring core framework (6/5/5/6/6/6) constructed from an indole ring and a cyclic diterpene moiety. Their structural variability mainly derived from the prenylation of indole ring and further modifications of isopentenyl groups such as oxidation and cyclization, as well as various modifications of the diterpenoid moiety. To date, more than 150 compounds within this class have been reported [8], and their fascinating structures and diverse bioactivities have made this class a hot topic in relevant scientific communities [9,10]. Recently, a series of new paspaline-type and its derived novel skeleton IDTs such as janthinellumines A–I [5], penpaxilloids A–E [7], and schipenindolenes A–H [6] with different biological activities were reported. Among them, schipenindolene A was found to be a potent HMG-CoA reductase degrader for lowering cholesterol, which was constructed by a concise and viable biomimetic semisynthesis [6]. In 2022, the Tong group reported the first asymmetric total syntheses of three paspaline-type IDTs paspalicine, paspalinine and paspalinine-13-ene, which provided a new synthetic strategy for the synthesis of other paspaline-type IDTs and would facilitate further investigation on these active IDTs [11].
Liver fibrosis was a pathological process induced by various chronic liver diseases, characterized by the excessive deposition and remodeling of the extracellular matrix (ECM) within the liver. This progressive condition not only disrupted the liver’s structural integrity and impaired its function but can also advance to liver cirrhosis, liver failure, and even hepatocellular carcinoma, posing a significant threat to patient health [12,13]. As a major global public health concern, liver fibrosis affected millions of individuals worldwide. However, there are no drugs specifically approved for the treatment of liver fibrosis on the market. Thus, the development of novel, low-toxicity, and highly effective antifibrotic agents is urgently needed.
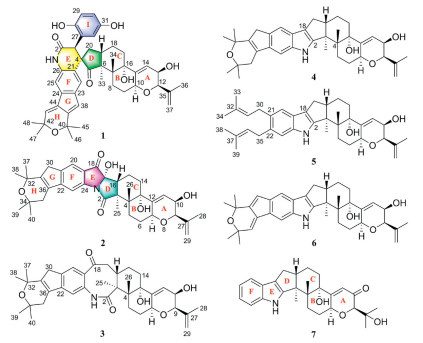
In our continuing search for Euphorbiaceae diterpenoids to treat liver fibrosis [14,15], the diverse structures and promising activities exhibited by IDTs produced by fungi had captured our attention. Therefore, uncovering its novel biological functions such as anti-hepatic fibrosis has become a paramount objective of our research. Guided by liquid chromatography-mass spectrometry (LC-MS)/MS-GNPS (Global Natural Product Social Molecular Networking) data, we analyzed the secondary metabolites produced by fungi and found the endophytic fungus Penicillium janthinellum H-6 isolated from the perennial herbaceous plant Euphorbia dracunculoides, which was a rich source of paspaline-type IDTs. As shown in Fig. 1, the results of screening against GNPS spectral database displayed two nodes with parent ions at m/z 585.35 and 480.181, which were identified as two paspaline-type IDTs, JBIR-137 and paxilline, respectively. Subsequently, further in-depth investigation of the rice fermentation of P. janthinellum H-6 led to the discovery of two unprecedented paspaline-derived IDTs (1 and 2) and their related biosynthetic precursors (3–7) (Fig. 2). Penispirolactam (1) represents a unique octacyclic skeleton (6/5/6/6/5/6/6/6) with a spiro core formed by the integration of a C6–C2 unit with the indole moiety, and penipyrroloindole (2) is a rearranged octacyclic skeleton (6/5/6/5/5/6/6/6) with a pyrrolo[1,2-a]indole core fragment. Herein, we reported the isolation, structural elucidation, putative biosynthetic pathways, and anti-liver fibrosis activation of 1–7.
Penispirolactam (1), a white amorphous powder, possessed a molecular formula of C45H51NO8 as deduced by high-resolution electrospray ionization mass spectrometry (HRESIMS) at m/z 734.3684 for [M + H]+ (calcd. 734.3688) and at m/z 756.3507 for [M + Na]+ (calcd. 756.3507), corresponding to 21 degrees of unsaturation. The infrared spectroscopy (IR) spectrum showed the presence of hydroxyl groups (3346 cm-1) and amide group (1659 cm-1). The 1H nuclear magnetic resonance (NMR) data of 1 (Table S1.1 in Supporting information) exhibited signals indicative of seven methyls [δH 1.00, 1.44, 1.44, 1.46, 1.495, 1.504, and 1.75 (each 3H, s)], five olefinic protons [δH 4.86 (1H, s), 5.07 (1H, s), 5.76 (1H, d, J = 6.0 Hz), 6.48 (1H, d, J = 1.6 Hz), and 6.92 (1H, d, J = 1.6 Hz)], three oxygenated methines [δH 3.77 (1H, s), 3.90 (1H, d, J = 6.0 Hz), and 4.53 (1H, overlapped)], a 1,3,4-trisubstitued phenyl [δH 6.34 (1H, d, J = 2.8 Hz), 6.46 (1H, dd, J = 8.6, 2.8 Hz), and 6.60 (1H, d, J = 8.6 Hz)], a 1,2,4,5-tetrasubstituted phenyl [δH 6.82 and 7.31 (each 1H, s)], and several aliphatic protons. The 13C NMR (Table S1.1) and DEPT spectra showed 45 carbon resonances, including a ketocarbonyl (δC 214.6), an amide carbonyl (δC 170.4), two benzene rings, a terminal double bond, three trisubstituted double bonds, seven methyls, five sp3 methylenes, five sp3 methines (three oxygenated ones), and six quaternary carbons (three oxygenated ones). As 14 of 21 degrees of unsaturation were ascribed to two phenyls, two carbonyls, and four double bonds, the remaining seven degrees of unsaturation suggested that 1 possessed a nine-rings system. Compared to the co-isolated known IDT, paxilline (7) [16], the aforementioned data shared most similarities with it, except for the additional signals for a C6-C2 unit, which suggested that 1 was a highly modified paspaline-type IDTs.
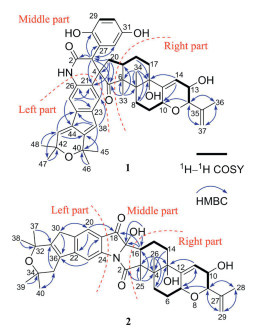
The planar structure of 1 was accomplished by 2D NMR data analysis (Fig. 3). The 1H–1H correlation spectroscopy (1H–1H COSY) correlations of H2-8/H2-9/H-10, H-12/H-13/H-14, and H2-17/H2-18/H-19/H2-20 indicated the presence of three fragments (C-8–C-9–C-10, C-12–C-13–C-14, and C-17–C-18–C-19–C-20), which connected with other groups were further determined by the interpretation of the key heteronuclear multiple bond correlation (HMBC) correlations. The HMBC correlations from H-12 and H-14 to C-10 and from H-13 to C-15 formed a partially hydrogenated pyran ring (A-ring). The HMBC correlations from H3-36 and H2-37 to C-12, along with H3-36 to C-37, indicated that an isopropenyl group was attached to C-12 of A-ring. The HMBC correlations from H2-8, H-14, and H2-18 to C-16, H3-34 to C-6, C-7, C-8, and C-16, and H3-33 to C-6, C-7, and C-19 connected these two fragments (C-8–C-9–C-10 and C-17–C-18–C-19–C-20) with the corresponding functional groups and the A ring to form a fused 6/6/6 tricyclic system. The structural feature of this tricyclic system in the right part of 1 was consistent with that of JBTR-137 (6) [17]. And in the left part of 1, by analyzing its NMR data (including the HMBC correlation shown in Fig. 3), it was determined that this part had the same 6/5/6 tricyclic structural fragment as 7. The middle part of compound 1 could be determined by combining HMBC correlations and degrees of unsaturation analysis. The HMBC correlations from H-3 to the C-27, C-28, and C-32 of the trisubstituted benzene ring and the HMBC correlations from H-32 to C-3 and H-3 to the amide carbonyl (C-2) supported the presence of the additional C6-C2 unit. The remaining one degree of unsaturation and the HMBC correlations from H-3 to C-5 and C-21, H2-20 to C-3, C-5, and C-21, H3-33 to C-5, and H-22 to C-4 confirmed the middle part of 1 was a spiral characteristic fragment formed by a δ-lactam and a cyclopentanone via the spiral carbon (C-4). Thus, the planar structure of 1 with an unprecedented spiro skeleton ring system constructed by the integration of a C6–C2 unit with the indole moiety was determined.
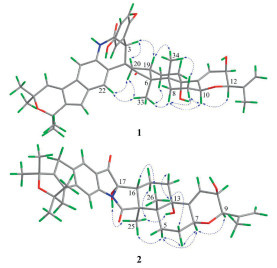
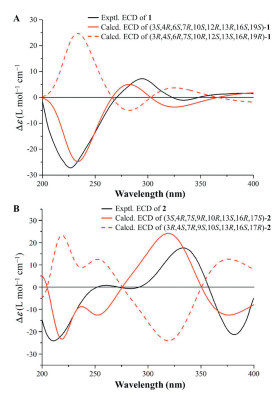
The relative stereochemistry of 1 was elucidated based on interpretation of nuclear overhauser effect spectroscopy (NOESY) interactions and 1H–1H coupling constants. The NOE cross-peaks of H-10/H-8α, H-10/H-9α, H-8α/H-9α, and H-9β/H3-34 as well as H3-34/H-19 and H-18/H3-33/H-8α indicated that both rings B and C were in a chair conformation and trans-fused (Fig. 4). Thus, the relative configurations of H-10, 16-OH, and CH3-33 were arbitrarily assigned α-orientations, while CH3-34 and H-19 were taking the β-orientations. The NOE correlations of H-10 with H-12 suggested H-12 was defined to be the α-orientation. The small coupling constant between H-12 and H-13 indicated these two protons was a cis-configuration. The relative configuration of the spiral carbon (C-4) could be determined as 4R* by the observed NOE correlations of H-22/H3-33, H3-33/H-20α, H-20α/H-22, H-19/H-3, and H-3/H-20β. Although H-3 showed a strong NOE correlation with H-20β and H-19, this information alone was insufficient to determine the relative configuration of C-3 (Fig. S1.1 in Supporting information). Therefore, two possible stereoisomers have been proposed: (3S*, 4R*, 6S*, 7R*, 10S*, 12R*, 13R*, 16S*, 19S*)-1a and (3R*, 4R*, 6S*, 7R*, 10S*, 12R*, 13R*, 16S*, 19S*)-1b (Fig. S1.2 in Supporting information). Subsequently, 1D NMR calculations based on gauge-independent atomic orbital (GIAO) theory were performed for both 1a and 1b scenarios, in combination with the experimental NMR shifts, were analyzed using the improved DP4+ probability methods. The final DP4+ score of (3S*, 4R*, 6S*, 7R*, 10S*, 12R*, 13R*, 16S*, 19S*)-1a (100.00%) afforded an absolute advantage over that of its counterpart (0.00%), allowing assignment of the 3S* of the C-3 (Fig. S1.2). Finally, the absolute configuration of 1 was determined by quantum-chemical calculations methods. Two possible conformational scenarios, (3S, 4R, 6S, 7R, 10S, 12R, 13R, 16S, 19S)-1a and its enantiomer, were subjected to theoretical electronic circular dichroism (ECD) calculations based on time-dependent density functional theory (TDDFT). As shown in Fig. 5A, the experimental ECD curve matches well with the calculated ECD curve of 1a, exhibiting the same first negative, second positive, and third negative Cotton effects around 227, 280, and 325 nm, respectively. Thus, the structure of compound 1 was unambiguously determined.
Penipyrroloindole (2) exhibited a molecular formular of C37H45NO7 with 16 degrees of unsaturation based on the HRESIMS data (m/z 638.3087 [M + Na]+, calcd. 638.3089). The 1H NMR data (Table S1.2 in Supporting information) indicated the presence of seven methyls [δH 0.89, 1.30, 1.32, 1.44, 1.44, 1.78, and 1.98 (each 3H, s)], three olefinic protons [δH 5.03 (1H, s), 5.18 (1H, s), and 5.80 (1H, d, J = 6.0 Hz)], three oxygenated methines [δH 3.85 (1H, d, br s), 3.95 (1H, d, J = 5.6 Hz), and 4.53 (1H, dd, J = 10.4, 8.8 Hz)], a 1,2,4,5-tetrasubstituted benzene ring [δH 7.57 and 7.66 (each 1H, s)], and a series of aliphatic protons. The 13C NMR (Table S1.3 in Supporting information) and DEPT spectra indicated 37 carbon resonances, which were classed as a conjugated ketocarbonyl (δC 197.7), an amide carbonyl (δC 179.4), a tetrasubstituted benzene ring, a terminal double bond, a trisubstituted double bond, a hydrogen-free double bond, seven methyls, six sp3 methylenes, four sp3 methines (three oxygenated), and six quaternary carbons including four oxygenated ones. The aforementioned information implied the existence of an octacyclic ring system in 2.
By analyzing the 1H–1H COSY and HMBC correlations of 2 (Fig. 3), it could be determined that its left and right parts were similar or identical to the left and right parts of 1, respectively. The similar left moiety was elucidated to be a 6/5/6 tricyclic system with a double bond at C-31 and C-36 by the presence of two isolated methylenes and one tetrasubstituted double bond instead of two conjugated double bonds as well as the HMBC correlations shown in Fig. 3. Subsequently, by combining the HMBC correlations and degrees of unsaturation analysis, it could be determined how the remaining three groups (the ketocarbonyl, amide, and oxygenated quaternary carbon) in the middle connect the left and right parts. The HMBC correlation from H-20 to the ketocarbonyl confirmed the conjugation of the ketocarbonyl group (C-18) with the benzene ring. The HMBC correlations from 17-OH to C-17 and C-16 (observed in DMSO‑d6) and H-16 to C-17 and C-18 confirmed the connection of C-18 and C-16 via the oxygenated quaternary carbon (C-17). The amide group was linked to C-3 supported by the HMBC correlation from H3-25 to the amide carbonyl (C-2). Since 14 of the 16 degrees of unsaturation were consumed by six rings (including one benzene ring), three double bonds, one ketocarbonyl group, and an amide carbonyl, the remaining two degrees of unsaturation implied that the N atom of the amide group (CONH2) must be connected to C-17 and C-24 to form two additional rings. Thus, the planar structure of 2 was defined to be a novel 6/5/6/5/5/6/6/6 octacyclic skeleton with a distinctive pyrrolo[1,2-a]indole moiety.
The relative configuration of 2 was determined by the NOESY data recorded in CDCl3 and DMSO‑d6. As shown in Fig. 4, the observed NOE correlations of H-9/H-7, H-7/H-5α, H-5α/H3-25, H3-25/H-15α, H-7/H-6α and H-6β/H3-26, H3-26/H-16, H3-26/H-14β in CDCl3 together with the small coupling constant of H-9 and H-10 suggested that the relative configurations of C-3, C-4, C-7, C-9, C-10, C-13, and C-16 were consistent with 6. Moreover, the significant NOE correlation of 17-OH/H3-25/13-OH observed in DMSO‑d6 undoubtedly confirmed the α-orientation of 17-OH and 13-OH in 2. The absolute configuration of 2 was assigned by comparison ECD spectrum with those calculated for two presumable isomers, (3S, 4R, 7S, 9R, 10R, 13S, 16R, 17S)-2 and (3R, 4S, 7R, 9S, 10S, 13R, 16S, 17R)-2. Based on the agreement between the experimental and calculated ECD curves (Fig. 5B), the stereochemistry of 2 was thereby assigned to be 3S, 4R, 7S, 9R, 10R, 13S, 16R, 17S.
Penijanidine A (3) presented the molecular formular C37H47NO6 with 15 degrees of unsaturation, by HRESIMS ([M + H]+, m/z 602.3470, calcd. 602.3476). The 1H and 13C NMR data of 3 (Tables S1.2 and S1.3) resembled those of the known paspaline-type IDTs, shearinie P [18]. The main differences were the carbonyl group at C-10 and the 2-hydroxyisopropyl group at C-9 in compound 3 had been converted to a hydroxyl group and an isopropenyl group, respectively. These changes were further confirmed by the 1H–1H COSY correlations of H-9/H-10/H-11 and the HMBC correlations from H3-28 and H2-29 to C-9 and C-27 (Fig. S1.3 in Supporting information). The cis-relationship of H-9 and H-10 were supported by its small coupling constant. The relative configuration of 3 was consistent with that of shearinine P according to comparable observations in the 1D NMR and NOESY experiments (Fig. S1.4 in Supporting information).
Penijanidine B (4) possessed the molecular formula of C37H47NO4 as established from the ion peak at m/z 570.3578 [M + H]+ (calcd. 570.3578) in its HRESIMS spectrum. The 1D NMR of 4 (Tables S1.2 and S1.3) closely resembled those of 6, except for the appearance of a tetrasubstituted double bond and two isolated methylenes in 4 replacing two conjugated trisubstituted double bonds in 6. This inferred that the 6/5 bicyclic ring formed by the diprenyl modification at C-21 and C-22 of the indole ring had different oxidation states. As shown in Fig. S1.3, the HMBC correlations from H3-37/38 to C-31, H3-39/40 to C-35, H-20 to C-30, and H2-35 and H-23 to C-36 supported that the tetrasubstituted double bond was located at C-31 and C-36 as well as the two isolated methylenes was placed at C-30 and C-35, respectively. Therefore, the structure of 4 was elucidated as shown.
Penijanidine C (5) displayed a sodium adduct HRESIMS ion observed at m/z 578.3612 (calcd. for C37H49NO3Na+, 578.3605), indicating the molecular formula C37H49NO3 with 14 degrees of unsaturation. Comparison of the 1D (Tables S1.2 and S1.3) and 2D NMR data (Fig. S1.3) revealed that 5 had the same paspaline skeleton as 6, with the only difference being that the two isopentenyl groups on its indole ring had not been oxidatively cyclized into a 6/5 bicyclic ring. The two independent prenyls at C-21 and C-22 could also confirmed by the 1H–1H COSY and HMBC correlations as shown in Fig. S1.3, as well as by analyses of the degrees of unsaturation. Thus, the structure of 5 was determined as shown.
Two known paspaline-type IDTs, JBIR-137 (6) [17] and paxilline (7) [16], were identified based on NMR data reported in the literature. Meanwhile, the structure of 7 was first confirmed by single-crystal X-ray diffraction (Fig. S1.5 in Supporting information).
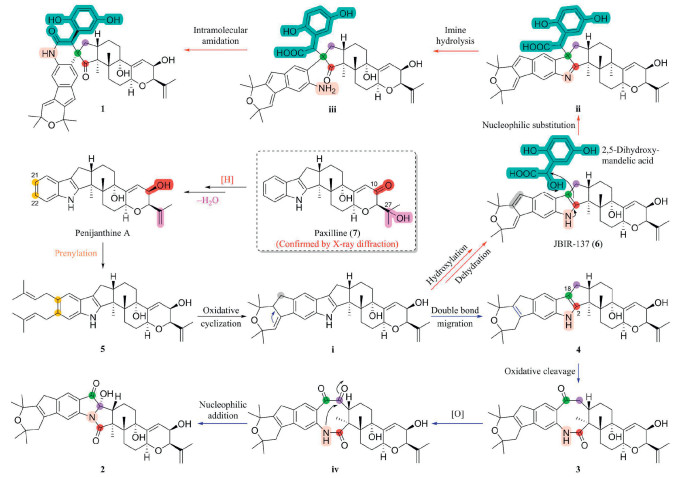
The structural diversity of the reported paspaline-type IDTs mainly lied in the structural modifications caused by prenylation on the indole ring and subsequent oxidation and cyclization of isopentenyl groups. And the obtained compounds 1 and 2 were two novel types of skeletons formed by ring opening, oxidation, rearrangement, and fusion of other fragments at the site where indole ring and diterpene were connected. To better understand these structures, the putative biosynthetic pathways for compounds 1 and 2 were proposed in Scheme 1. This proposal was developed on the basis of the co-isolation of compounds 3–7, which could be considered as the potential biosynthetic precursors or key intermediates. Initially, the ketocarbonyl (C-10) and the 27-OH of paxilline (7) underwent reduction and elimination reactions, respectively, to generate penijanthine A, and followed by the prenylation of penijanthine A at C-21 and C-22 to produce the key intermediate penijanidine C (5). Subsequently, the oxidative cyclization of the two isopentenyls of 5 could form the key intermediate ⅰ (Scheme S1.1 in Supporting information) [9], which underwent hydroxylation followed by dehydration or double bond migration to obtain JBIR-137 (6) and penijanidine B (4), respectively. Utilizing these two compounds as intermediates, new skeleton types of IDTs penispirolactam (1) and penipyrroloindole (2) might be formed through different pathways. The first pathway involved a nucleophilic substitution reaction between compound 6 at C-18 position and the natural product 2,5-dihydroxymandelic acid to realize the integration of the C6–C2 fragment for generation of the key intermediate ⅱ. Furthermore, hydrolysis of the imine bond in intermediate ⅱ generated the intermediate ⅲ, and followed by intramolecular amidation to form 1. For another pathway, by oxidative cleavage of the Δ2,18 double bond of 4, new compound penijanidine A (3) were generated, which was then oxidized and underwent intramolecular nucleophilic addition reaction to ultimately form the novel skeleton IDT, penipyrroloindole (2).
The anti-liver fibrosis activity of compounds 1–7 was evaluated by Western blot (WB) analysis in transforming growth factor-β1 (TGF-β1)-stimulated LX-2 cells (hepatic stellate cells), a widely utilized cellular model for liver fibrosis research [19]. By analyzing the inhibitory effect of compounds on fibrosis-related biomarkers such as fibronectin, collagen I, and α-smooth muscle actin (α-SMA), potential active compounds for anti-liver fibrosis can be quickly identified [20]. And the TGF-β signaling inhibitor, SB431542 (SB), was used as the positive drug [21,22]. As shown in Fig. 6A, TGF-β1 stimulation significantly increased the production of fibronectin, collagen I, and α-SMA in cells, which could be reversed by treatment with compound 1 at 10 µmol/L. Overall, compound 1 exhibited the most outstanding inhibitory efficiency. The anti-liver fibrosis activity of compound 1 was further substantiated through immunofluorescence staining and concentration-dependent assays (Figs. 6B and C). Additionally, no significant cytotoxicity of all compounds was observed at the effective concentrations (Fig. S1.6 in Supporting information). Therefore, the active compound 1 was selected for further mechanism research.
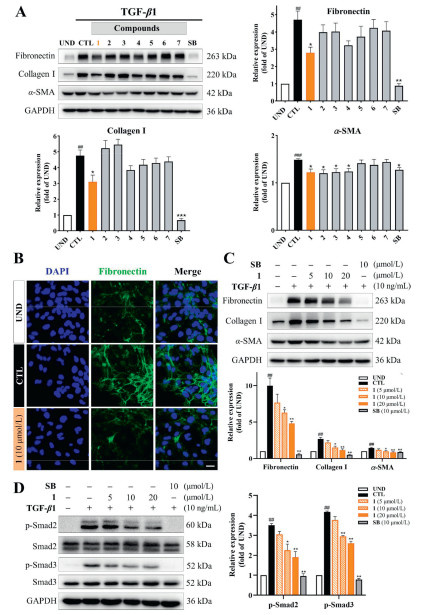
It is well known that the TGF-β/Smad signaling pathway is a well-established fibrosis-related signaling pathway. This pathway primarily involves the phosphorylation of Smad2 and Smad3, followed by the formation of complexes and translocate into the nucleus to promote the expression of extracellular matrix proteins, thereby driving fibrosis progression [23,24]. Subsequently, the impact of the active compound on the levels of p-Smad2 and p-Smad3 as well as Smad2 and Smad3 were analyzed on this model. As illustrated in Fig. 6D, stimulation with TGF-β1 markedly increased the phosphorylation levels of Smad2 and Smad3. While compound 1 dose-dependently reduced the levels of p-Smad2 and p-Smad3 without affecting the total protein levels of Smad2 and Smad3. These findings suggested that compound 1 exerted its anti-fibrotic effects by inhibiting the TGF-β/Smad signaling pathway (Fig. S1.7 in Supporting information).
In conclusion, five new IDTs, penispirolactam (1), penipyrroloindole (2), and penijanidines A–C (3–5), together with two known ones, JBIR-137 (6) and paxilline (7), were obtained from the endophytic fungus P. janthinellum H-6 guided by LC-MS-based molecular network. The two novel skeleton types of IDTs, penispirolactam and penipyrroloindole, could be regarded as derived from the co-isolated paspaline-type IDTs, 3–7, through different pathways. Moreover, the investigation on the anti-hepatic fibrosis functions of IDTs was carried out to further expand their pharmaceutical application. The activity assays indicated that compound 1 could exert its anti-hepatic fibrosis effects by inhibiting the TGF-β/Smad signaling pathway without cytotoxicity. Therefore, IDTs with novel skeletons were expected to develop into new chemical entities for low toxicity and high efficiency anti liver fibrosis drugs.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Lei Li: Writing – original draft, Methodology, Investigation. Xin-Ying Zhu: Writing – original draft, Methodology, Investigation. Jun-Yu Zhu: Methodology, Investigation. Zhi-Hao Wu: Methodology. Yan-Jiang Zhang: Methodology. Fang-Yu Yuan: Methodology, Funding acquisition. Dong Huang: Methodology, Funding acquisition. Sheng Yin: Writing – review & editing, Supervision, Resources, Funding acquisition. Gui-Hua Tang: Writing – review & editing, Supervision, Resources, Project administration, Funding acquisition.
This work was supported by the National Natural Science Foundation of China (Nos. 82273804, 22407144, and 82404454), the Science and Technology Program of Guangzhou, China (No. 2024B03J1322), the Science and Technology Planning Project of Guangdong Province, China (No. 2023A1111120025), and the Open Program of Shenzhen Bay Laboratory (No. SZBL2021080601007, China). Cartoons in the Schematic diagram (Fig. S1.7) were created with BioRender.
Supplementary material associated with this article can be found, in the online version, at doi:
T. Rodrigues, D. Reker, P. Schneider, et al., Nat. Chem. 8 (2016) 531–541. doi: 10.1038/nchem.2479
Y. Wang, P. Tang, W. Tu, et al., Chin. Chem. Lett. 36 (2025) 109955. doi: 10.1016/j.cclet.2024.109955
J.G. Ondeyka, G.L. Helms, O.D. Hensens, et al., J. Am. Chem. Soc. 119 (1997) 8809–8816. doi: 10.1021/ja971664k
W. Yang, T. Chen, Q. Tan, et al., J. Nat. Prod. 86 (2023) 1392–1401. doi: 10.1021/acs.jnatprod.2c01172
F. Cao, X.M. Liu, X. Wang, et al., Bioorg. Chem. 141 (2023) 106863. doi: 10.1016/j.bioorg.2023.106863
X.Z. Su, L.F. Zhang, K. Hu, et al., Angew. Chem. Int. Ed. 63 (2024) e202313859. doi: 10.1002/anie.202313859
L.T. Dai, L. Yang, J.C. Guo, et al., Bioorg. Chem. 145 (2024) 107205. doi: 10.1016/j.bioorg.2024.107205
J.W. Niu, J.Z. Qi, P.C. Wang, et al., Nat. Prod. Bioprospect. 13 (2023) 3. doi: 10.1007/s13659-022-00368-7
C. Liu, A. Minami, T. Dairi, et al., Org. Lett. 18 (2016) 5026–5029. doi: 10.1021/acs.orglett.6b02482
K. Tagami, C. Liu, A. Minami, et al., J. Am. Chem. Soc. 135 (2013) 1260–1263. doi: 10.1021/ja3116636
L.D. Guo, Z. Xu, R. Tong, Angew. Chem. Int. Ed. 61 (2021) e202115384.
M. Parola, M. Pinzani, Mol. Asp. Med. 65 (2019) 37–55. doi: 10.1016/j.mam.2018.09.002
D.Y. Zhang, S.L. Friedman, Hepatology 56 (2012) 769–775. doi: 10.1002/hep.25670
S. Li, L. Gan, Y.J. Tian, et al., Bioorg. Chem. 114 (2021) 105222. doi: 10.1016/j.bioorg.2021.105222
L. Gan, Q. Jiang, D. Huang, et al., Nat. Chem. Biol. 21 (2024) 80–90.
C. Matsui, Y. Ikeda, H. Iinuma, et al., J. Antibiot. 67 (2014) 787–790. doi: 10.1038/ja.2014.63
T. Kawahara, A. Nagai, M. Takagi, et al., J. Antibiot. 65 (2012) 535–538. doi: 10.1038/ja.2012.64
L.M. Zhou, F.D. Kong, P. Fan, et al., J. Nat. Prod. 82 (2019) 2638–2644. doi: 10.1021/acs.jnatprod.9b00620
L. Xu, A.Y. Hui, E. Albanis, et al., Gut 54 (2005) 142–151. doi: 10.1136/gut.2004.042127
E. Altrock, C. Sens, C. Wuerfel, et al., J. Hepatol. 62 (2015) 625–633. doi: 10.1016/j.jhep.2014.06.010
G.J. Inman, F.J. Nicolas, J.F. Callahan, et al., Mol. Pharm. 62 (2002) 65–74. doi: 10.1124/mol.62.1.65
J. Zhang, R. Li, Q. Liu, et al., Mol. Pharm. 17 (2020) 4152–4162. doi: 10.1021/acs.molpharmaceut.0c00633
F. Xu, C. Liu, D. Zhou, et al., J. Histochem. Cytochem. 64 (2016) 157–167. doi: 10.1369/0022155415627681
J. Massagué, Nat. Rev. Mol. Cell Biol. 13 (2012) 616–630. doi: 10.1038/nrm3434

Figure 1 LC-MS-based molecular networking of the EtOAc extract of fungus P. janthinellum H-6.

Figure 6 Identification of 1 as a potent antifibrotic compound in TGF-β1-stimulated LX-2 cells. (A) The effects of 1–7 on fibronectin, collagen I, and α-SMA. WB analysis was carried out in cells treated with vehicle or 10 µmol/L compounds for 48 h (n = 3 per group). (B) Immunostaining of fibronectin (green) in LX-2 cells. TGF-β1-activated LX-2 cells were treated with vehicle or 10 µmol/L compound 1 for 48 h. Scale bar: 20 µm. (C) Compound 1 suppressed the protein levels of fibronectin, collagen I, and α-SMA in a dose-dependent manner in TGF-β1-stimulated LX-2 cells. Cells were treated with vehicle or indicated concentrations of 1 for 48 h (n = 3 per group). (D) Influence of 1 on expressions of Smad2, Smad3, p-Smad2, and p-Smad3 in TGF-β1-stimulated LX-2 cells treated with indicated concentrations of 1 or SB for 48 h. CTL, cells treated with TGF-β1; UND, cells treated with vehicle; SB, SB431542. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. Data are presented as means ± SEM (A, C, D). ##P < 0.01, ###P < 0.001 vs. UND group; *P < 0.05, **P < 0.01 vs. CTL group.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:





 下载:
下载: