
Scheme 1.
Synthesis of metallopolymer microgels.
Light-mediated CO2-responsiveness of metallopolymer microgels
Xiaofei Wang , Xuezhen Lin , Huijuan Qiu , Jianda Xie , Zhengyu Lu , Yusong Wang , Weitai Wu

Stimuli-responsive polymeric materials that can reversibly respond to CO2 provides great opportunities [1-6]. Besides the prospect of CO2 capture and removal in easing the world's energy and environmental problems, with CO2 as a stimulus to induce polymer phase transitions, it is able to regulate properties of CO2-responsive polymeric materials for cell mimics, drug delivery, and many other applications [6-12].
Because of the prospective applications, efforts have been made to explore fundamental principles of polymer design to enable CO2-responsiveness, including the choice of CO2-responsive moieties and how effective CO2-responsiveness can be expected to be under the conditions of interest [11, 12]. Up to now, CO2-responsive polymeric materials are principally synthesized by the functionalization of polymer chains with basic groups, which can react with acidic CO2 in water based on acid–base pair theory [7-11]. Owing to reversible nature of the acid–base equilibrium, CO2 can be removed by bubbling with inert gases (e.g., N2 gas) under a mild condition. This offers an advantage of free of contamination by accumulated chemicals; yet, it also makes the materials barely respond to dilute CO2, due to low capacity of the CO2-responsive polymeric materials for CO2 capture from dilute CO2 sources, which undisputedly is a critical drawback [1-12]. As a theoretical possibility, enhancing CO2 binding affinity might allow CO2 capture of high capacity [13-16], which paves the way to address this problem [11] and thus have recently received attention in CO2-responsiveness design [17, 18]. For instance, efforts are made by exploiting frustrated Lewis pair (FLP) [19], and use CO2 to bridge FLP-containing polymers via dynamic covalent bonding [17, 18]. However, high CO2 binding affinity in turn leads to difficulty in CO2 removal without heating (≥ 60 ℃ typically). The challenge of seeking stimuli-responsive polymeric materials of a distinct mechanism to allow both CO2 capture of high capacity and CO2 removal of good reversibility under a mild condition remains to be solved.
In this work, we would like to report a concept of how to tune CO2 binding affinity to mediate CO2-responsiveness by using side-chain type metallopolymers containing metalla-aromatics as CO2-responsive moieties on microgels. Unlike bases or FLP groups reported in previous articles that give rise to intra-/intermolecular acid-base activation of CO2 via ionic or dynamic covalent bonding [1-14], metallopolymers (or organometallic polymers) with metal functionalities display typically high CO2 binding affinity via strong interactions (e.g., metal complexes) [20-22], which however are rarely used for harnessing CO2-responsiveness. Here our intention to use those metallopolymers containing metalla-aromatics is because most metalla-aromatics are thermodynamic stable and possess properties of both organometallics and aromatics [23, 24]. An alteration in (anti)aromaticity is known to efficiently tune reactivity [25]. For instance, (anti)aromaticity can promote reactivity of FLP groups with small molecules like CO2 [26, 27]. Interestingly, light exposure could lead to a change in (anti)aromaticity, offering a facile way to vary reactivity [28-30]. This tunning in reactivity may depend on a combination of properties: the number of electrons, the orbital topology, and the electronic state, in the sense that a change in each of these factors lead to a reversal in the allowedness and forbiddenness; in particular, the results pointed to a reversal of (anti)aromaticity following photoexcitation [28-30]. Inspired by these, one wonders if it is possible to obtain polymeric materials, microgels [3, 31-33] here of metallopolymers containing metalla-aromatics, of effective and reversible CO2-responsiveness by utilizing light as a mediator (Scheme 1).
The microgels were synthesized through electrochemical polymerization [34] of a Craig-type coplanar Möbius metalla-aromatic compound (OsHEMA; 2.0 × 10−4 mol/L) [35] with a crosslinker N, N'-methylenebisacrylamide (MBAAm; 2.0 × 10−6 mol/L), and with copper nanoparticles (36 ± 5 nm; see Fig. S1 in Supporting information for TEM, XRD and UV-vis absorption results) as mediators for electron transfer, so as to avoid direct electrolysis of OsHEMA on the electrode that might lead to disproportional reactions [36]. On an anodic scan, an oxidation starts to develop on copper nanoparticles at a low potential of ca. +0.2 V versus Ag/AgCl (Fig. S2 in Supporting information), leading to formation of strong oxidizing copper species [36, 37], which can attack MBAAm to form N-localized radicals [34, 36]. These N-localized radicals could trigger a cascade of reactions that typically involve the loss of a proton and formation of C-localized radicals, which then initiate polymerization on attacking vinyl groups of the monomer/crosslinker, making them enter into free radical polymerization [36] and particle growth processes in the electrochemical systems [34, 38]. In this circumstance, the synthesis should proceed upon applying a suitable potential of +0.3 V that is above the oxidation wave of copper nanoparticles, but below those of OsHEMA (starting at ca. +0.6 V versus Ag/AgCl) and MBAAm (starting at +1.6 V versus Ag/AgCl) (Fig. S3 in Supporting information), resulting in toroidal-like microgels (collected after reacting for 4 h at 25.0 ℃, and purified before characterization; Fig. 1a). The mean diameter of freeze-dried toroids is 1.46 ± 0.21 µm, and the width is 69 ± 23 nm; the outline is barely sharp due to the existence of corona [34, 39] on surface that may be indistinctly seen in Fig. 1b. Energy dispersive X-ray (EDX) elemental mapping results indicated the existence of carbon, oxygen, phosphorus and osmium in an individual toroid; no copper salts or other inorganics were detected (Figs. 1c-f). The edge of EDX elemental maps matches well that in annular dark-field TEM image (Fig. 1b), indicating that those elements and corresponding moieties are distributed throughout microgels. 31P NMR spectrum of the microgels (Fig. S4 in Supporting information) showed two signals of the metalla-aromatic moiety at ca. 13.0 (for CPPh3) and 3.6 ppm (for OsPPh3), and in 1H NMR spectrum (Fig. S5 in Supporting information) at ca. 12.2 (for OsCH) and 7.1-8.2 ppm (for other aromatic protons) [35], indicating the integration of the metalla-aromatic moiety to the microgels. The weight percentage of the metalla-aromatic moiety was estimated to be ca. 95.4 wt%, via analyzing the percentage of osmium metal by inductively coupled plasma mass spectrometry [35], which was close to the feeding for microgel synthesis (ca. 99.9 wt%). The microgels were hydrophilic (Fig. S6 in Supporting information) and kept stable in water (Fig. S7 in Supporting information). To verify the shape and composition of microgels (Table S1 in Supporting information), over five samples were repeated, and ten samples were prepared at 20–30 ℃, which displays negligible influence on synthesis. The microgels were reproducible from batch to batch, with an average yield of ≥ 80%.

Phase diagrams for aqueous microgel dispersion (0.015 wt%) were studied by UV-vis spectroscopy at a wavelength λ = 700 nm (of incident light intensity I0), where absorption is negligible (Fig. S8 in Supporting information). The decrease in transmitted light intensity (IT) can be associated with scattering, to give the normalized attenuation coefficient δλ as [34]:
|
|
(1) |
where the light attenuation Dλ = lg(I0/IT), c for the microgel concentration (wt%), and d for the cuvette thickness (1 cm). The tests were made at a slow temperature ramp (3 ℃/h; using a temperature controller, ±0.1 ℃) followed by a long waiting (30 min, after temperature reaching equilibrium) and a rapid measurement time (< 1 s), to ensure in the equilibrium state of microgels at each measurement. The dispersion was bubbled with N2 or CO2 gas (20.0 mL/min under 1 atm in dark) and equilibrated in a cuvette for tests. Upon purge with CO2 at a set temperature in the range of 20–80 ℃, the δ700 changed immediately and reached equilibrium within 30 min of CO2 purge (Fig. S9 in Supporting information), and the δ700 (equilibrated; the same below) increased with the amount of CO2 going through the dispersion via adjusting purge time (Fig. 2a). The increase in the δ700 is possibly associated with the binding of CO2 to the metalla-aromatic moiety [20-22], and ether oxygens as well [33], which could reduce hydrogen bonding of polymers with water and thus the hydrophilicity that lead to the drop of transmittance [34]. Since this subtle variation can also disrupt counterbalance of hydrophilic and hydrophobic forces in microgels, which is a key factor responsible for thermo-responsiveness [39, 40], then, temperature-dependence on the δ700 was studied. Results indeed show an increased δ700 at elevated temperatures (Fig. 2a), representing a possible transition at ca. 54.6 ℃ upon N2 purge (Fig. S10 in Supporting information); the transition temperature drops to ca. 47.5 ℃, 43.3 ℃ and 41.8 ℃, respectively, after a 30 s, 5 min and 30 min of CO2 purge (Figs. S10 and S11 in Supporting information). Those results demonstrate that the microgels are CO2-responsive. In further cyclic tests (Fig. 2b and Fig. S9), purge again with N2 following the CO2 purge, the δ700 cannot fully recover. The recovery ratio Rδ is ca. 5% at 20.0 ℃, as estimated by Rδ = (δCO-δN)/(δCO-δ0), in which δ0 is the initial δ700, and δCO is the δ700 after a CO2 purge and δN is that after one cycle of CO2/N2 purge. A larger Rδ appears at the higher temperature (Fig. 2b and Fig. S12 in Supporting information). However, unsatisfying recovery ratios were recorded over the range of temperatures for turbidity analysis (20–80 ℃; e.g., Rδ ≈ 45% at 80.0 ℃), demonstrating a poor reversibility.
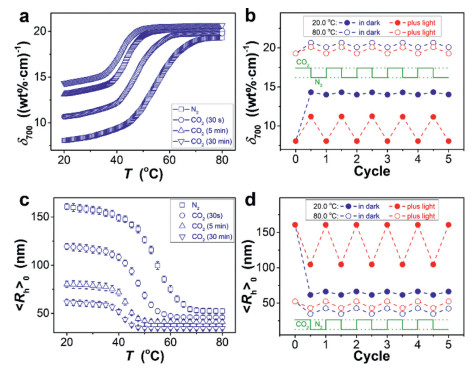
The reversibility of CO2-responsiveness can be improved by irradiation to an ultraviolet lamp (offering 30.0 W/cm2 area light of a wavelength 365 nm) (Fig. 2b; keeping the temperature at the set value while turning off gas purge and the lamp for rapid measurements, the same below). This seems to be realized by altering the variations on the δ700 under CO2/N2 purge. Compared with results measured in dark, when the light was applied, the rising extent of the δ700 upon purge with CO2 became lower, whereas the reducing extent of the δ700 became more significant upon purge again with N2. Taken together the two variation manners, light-mediated CO2-responsiveness could be achieved and the Rδ approached 100% at 20.0 ℃, indicating good reversibility under a mild condition. Similar results were obtained at other temperatures in the range of 20–80 ℃ (e.g., at 80.0 ℃, Fig. 2b and Fig. S12).
An examination on the observed δ700 change may raise a few questions. The first possible question associates with aggregation: may the recorded δ700 relate to aggregation of microgels? It has been reported that macromolecular clews in solutions below a critical concentration (ca. 0.1 wt%) can conserve individuality [41]. Given that the δ700 was recorded on dispersions of a much lower concentration (0.015 wt%), aggregation of microgels may be excluded. This hypothesis is supported by particle dynamics, through characterizing particle diffusion using an exponent P that is obtained by diffusing wave spectroscopy. If aggregation occurred, a change on particle dynamics can be reflected in a change from freely diffusive (P ≈ 1) to subdiffusive motion (P < 0.8) [42]. Here, a constant P of ca. 1 is obtained in the temperature range of 20–80 ℃ (Fig. S13 in Supporting information), confirming that the microgels did not lose individuality. The δ700 change reflects transitions of individual microgels. To provide a direct evidence, dynamic light scattering (DLS) was used to monitor the average hydrodynamic radius < Rh > 0, which is "equivalent sphere radius" [43, 44] that is obtained by angular extrapolation of apparent diffusion coefficient of microgels. As expected, the equilibrated < Rh > 0 decreased as CO2 purge time increasing (Fig. 2c), rendering a large volumetric swelling ratio SW(N2/CO2) = (< Rh > 0(initial, N2)/ < Rh > 0(after a 30 min of CO2 purge)) [3] of 17.9 at 20.0 ℃ in dark, indicating a high CO2-responsivity. For each case of the same CO2 purge time, the microgels deswelled at elevated temperatures until a sharp but continuous change around the δ700 transition temperature (Fig. S14 in Supporting information), resulting in a smaller difference between < Rh > 0 under N2/CO2 purge at the higher temperature (e.g., SW(N2/CO2) = 3.5 at 80.0 ℃). When the light was applied, full reversible CO2-responsiveness was also observed (Fig. 2d), indicating existence of light-mediated CO2-responsiveness on individual microgels.
The second possible question relates to the observation of light-mediated CO2-responsiveness of the microgels. The characteristics of the change in δ700 and < Rh > 0 upon CO2/N2 purge in light is highly reminiscent of consequences of photoexcitation [45-47], which has been reported previously on metallopolymers that the internal energy absorbed from an exciting radiation could be lost by nonradiative processes among others [23, 35]. Internal conversion (driven by the kinetic coupling between electronic states; Fig. S15 in Supporting information) and the intersystem crossing (mediated by the spin–orbit coupling) are main intramolecular nonradiative processes [45-47]. Note that most organics following photoexcitation undergo rapid internal conversion to the lowest excited singlet state (S1) [46, 47], but the internal conversion from S1 to the lowest singlet state S0 is relatively slow, allowing time to promote intersystem crossing rapidly of S1-state to the lowest triplet state T1 of organometallics via heavy-atom effect without significantly perturbing the excited-state dynamics [48], which facilitates formation of T1-state of metallapentalenes [24, 49]. As singlet and triplet states are distinct in nature [45], particularly (anti)aromaticity [28-30, 47], the above explanation should be generalized for light-mediated CO2-responsiveness of the microgels. In a density functional theory (DFT) calculation, metallapentalene with osmium center and with a strong π-acceptor ligand (like CO; Scheme 1) was proposed to be aromatic in S0-state, whereas antiaromatic in T1-state [49]. (Anti)aromaticity refers to multidimensional manifestation in energetics, reactivity, electron delocalization and other properties, all of which in turn have been used to define numerous indices of (anti)aromaticity [24, 28-30]. For instance, the gain of aromaticity in FLP groups for the activation of asmall molecule (e.g., CO2) may result in a gain of stability in both the corresponding transition state (leading to a lower barrier transformation), and in final adduct (making the process thermodynamically more favorable) [26, 27]. Thereby, it appears that light-mediated CO2-responsiveness is driven by the loss of aromaticity from initial photoexcitation and concurrent formation of a less reactive, antiaromatic excited state of relatively low CO2 binding affinity, and by subsequent relief of antiaromaticity that can enhance CO2 removal upon N2 purge in light, that is, the origination of good reversibility. In comparison with results recorded at the same temperature in dark, the decrease in δ700 (Fig. 2b) and increase in < Rh > 0 (Fig. 2d) upon CO2 purge in light should support the suppression of light for CO2 binding, thus, indirectly reflect the feasibility of the explanation.
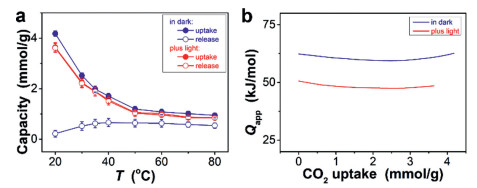
Given that the difference in CO2 binding affinity can lead to a change in CO2 uptake/release capacity [13-17], single-gas uptake-release experiments were performed. Fig. 3a indicates a larger CO2 uptake capacity of the microgels in dark than that acquired in light at the same temperature of 20.0 ℃, in agreement with the variation in the δ700 and < Rh > 0 upon CO2 purge between in dark and in light. Experiments at other temperatures in the range of 20–80 ℃ and over a pressure range from 0.1 atm to 1 atm indicated similar results (Fig. S16 in Supporting information). These results also demonstrate the dominate impact of the intersystem crossing, although the influence of the heat generated by internal conversion is ineluctable. Based on those isotherms (Fig. S17 in Supporting information), apparent isosteric heat of CO2 uptake (Qapp) for the microgels was estimated (Fig. 3b) [50]. The reduction on the Qapp was apparent on light exposure (e.g., Qapp ≈ 50.6 kJ/mol at zero loading and at 20.0 ℃), compared to that in dark (Qapp ≈ 62.4 kJ/mol at zero loading at 20.0 ℃). It is reasonable that light had acted as a mediator to tune CO2 binding affinity [13-16]. This is further supported by CO2 removal ability (Fig. 3a). Over 95% of the CO2 captured in light was released by N2 purge also in light at the same temperature, standing in contrast against barely or partial (< 54%) release in case of uptake/release in dark (Fig. S18 in Supporting information). The observation is consistent with turbidity and DLS results, well connecting light-mediated CO2-responsiveness to CO2 uptake/release capacity.
Further optimizing the experiments via a combination of CO2 capture in dark and CO2 removal in light, the former can enable a large CO2 uptake capacity of ca. 4.2 mmol/g under 1 atm CO2 pressure at 20.0 ℃, and the latter allows partial release (by keeping CO2 purge while exposing to light) or full release (by N2 purge in light) of CO2 under ambient pressure at 20.0 ℃ (Fig. 4a). This finding is curious, since both CO2 capture of high capacity and CO2 removal of good reversibility are realized under a mild condition, without large temperature swings (Fig. S19 in Supporting information) that is required for CO2 uptake/release reported previously (Table S2 in Supporting information) [1–18]. It is the change in the heat of uptake that produces large differences in binding of CO2 to polymers: CO2 capture in dark favors the formation of strong interactions (the Qapp above 60 kJ/mol; e.g., metal complexes), and light exposure facilitates weak interactions (the Qapp below 60 kJ/mol; e.g., quadrupole-π) coming to dominate [20, 51, 52]. As strong interactions favor CO2 capture from dilute sources, one may wonder how low a CO2 level can induce an effective CO2-responsiveness. To this end, the dispersion was bubbled with CO2/N2 mixed-gases (Fig. 4b). Distinctly, the microgels could respond to CO2 at low CO2 levels, as evidenced by attainment of a plateau of CO2 uptake capacity ≥3.9 mmol/g even with CO2 level lower down to 30 vol% in dark (Fig. 4c); correspondingly, the microgels deswelled, with < Rh > 0 ≤ 68.2 nm (Fig. 4d) or SW(N2/CO2) ≥ 13.1 (Fig. S20 in Supporting information). If we defined the effective CO2-responsiveness as a CO2 level under which a 10% < Rh > 0 change was measured, the critical CO2 level was ca. 1 vol% in dark, which increased to ca. 70 vol% in light. This also indicates that, for the mixed-gas containing 1–70 vol% CO2, the microgels could effectively respond in dark, and nearly recover by simply exposing to light.

Light-mediated CO2-responsiveness was also observed on spherical metallopolymer microgels (Fig. S21 in Supporting information). These spherical microgels could also exhibit both CO2 capture of high capacity (ca. 3.9 mmol/g under 1 atm at 20.0 ℃ in dark) and CO2 removal of good reversibility (by exposing to light) under a mild condition (Fig. S22 in Supporting information).
In summary, we demonstrate the concept of mediating CO2-responsiveness through tuning CO2 binding affinity by using side-chain type metallopolymers containing metalla-aromatics as CO2-responsive moieties on microgels. Studies on the dilute aqueous dispersions of these microgels in dark indicate high CO2-responsivity, but poor reversibility upon N2 purge, which can be improved by applying light exposure. This light-mediated CO2-responsiveness can be elucidated by the loss of aromaticity from initial photoexcitation and concurrent formation of a less reactive, antiaromatic excited state of relatively low CO2 binding affinity, and by subsequent relief of antiaromaticity that can enhance CO2 removal. Our results underscore the vast potential of bridging metalla-aromatic chemistry and polymer science to enable both CO2 capture of high capacity and CO2 removal of good reversibility under a mild condition, providing a novel strategy for the design of valuable materials that can effectively, reversibly respond to dilute CO2.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work is supported by National Natural Science Foundation of China (Nos. 21774105, 21805164, 20923004), Chuying Plan Youth Top-notch Talents of Fujian Province, and National Fund for Fostering Talents of Basic Science (No. J1310024).
Q. Yan, R. Zhou, C.K. Fu, et al., Angew. Chem. Int. Ed. 50 (2011) 4923-4927. doi: 10.1002/anie.201100708
D.H. Han, X. Tong, O. Boissière, Y. Zhao, ACS Macro Lett. 1 (2012) 57-61. doi: 10.1021/mz2000175
H. Yu, K. Imamura, M. Yue, G. Inoue, Y.Y. Miura, J. Am. Chem. Soc. 134 (2012) 18177-18180. doi: 10.1021/ja3080192
K.C. Jie, Y. Yao, X.D. Chi, F.H. Huang, Chem. Commun. 50 (2014) 5503-5505. doi: 10.1039/c4cc01704h
K.C. Jie, Y.J. Zhou, Y. Yao, B.B. Shi, F.H. Huang, J. Am. Chem. Soc. 137 (2015) 10472-10475. doi: 10.1021/jacs.5b05960
K. Jie, Y. Zhou, Y. Yao, B. Shi, F. Huang, J. Am. Chem. Soc. 137 (2015) 10472-10475. doi: 10.1021/jacs.5b05960
A. Darabi, P.G. Jessop, M.F. Cunningham, Chem. Soc. Rev. 45 (2016) 4391-4436. doi: 10.1039/C5CS00873E
Q. Zhang, L. Lei, S.P. Zhu, ACS Macro Lett. 6 (2017) 515-522. doi: 10.1021/acsmacrolett.7b00245
H.B. Liu, S.J. Lin, Y.J. Feng, P. Theato, Polym. Chem. 8 (2017) 12-23. doi: 10.1039/C6PY01101B
B.X. Jiang, Y.C. Zhang, X.D. Huang, et al., Ind. Eng. Chem. Res. 58 (2019) 15088-15108. doi: 10.1021/acs.iecr.9b02433
M.F. Cunningham, P.G. Jessop, Macromolecules52 (2019) 6801-6816. doi: 10.1021/acs.macromol.9b00914
S.Y. Wang, Q.H. Liu, L. Li, M.W. Urban, Macromol. Rapid Commun. (2021) 2100054. doi: 10.1002/marc.202100054
B.S. Zheng, J.F. Bai, J.G. Duan, L. Wojtas, M.J. Zaworotkoet, J. Am. Chem. Soc. 133 (2011) 748-751. doi: 10.1021/ja110042b
F. Ding, X. He, X.Y. Luo, et al., Chem. Commun. 50 (2014) 15041-15044. doi: 10.1039/C4CC06944G
X.Y. Luo, X.Y. Chen, R.X. Qiu, et al., Dalton Trans. 48 (2019) 2300-2307. doi: 10.1039/c8dt04680h
P. Das, S.K. Mandal, Chem. Mater. 31 (2019) 1584-1596. doi: 10.1021/acs.chemmater.8b04683
L. Chen, R.J. Liu, Q. Yan, Angew. Chem. Int. Ed. 57 (2018) 9336-9340. doi: 10.1002/anie.201804034
L. Chen, R.J. Liu, X. Hao, Q. Yan, Angew. Chem. Int. Ed. 58 (2019) 264-268. doi: 10.1002/anie.201812365
D.W. Stephan, G. Erker, Angew. Chem. Int. Ed. 54 (2015) 6400-6441. doi: 10.1002/anie.201409800
M. Aresta, A. Angelini. The carbon dioxide molecule and the effects of its interaction with electrophiles and nucleophiles, in: X.B. Lu (Ed. ), Carbon Dioxide and Organometallics, Vol. 53, Springer Nature Switzerland AG, 2016, pp. 1-38.
Y.L. Wang, D. Astruc, A.S. Abd-El-Aziz, Chem. Soc. Rev. 48 (2019) 558-636. doi: 10.1039/C7CS00656J
E.J. Kim, R.L. Siegelman, H.Z. Jiang, A. C. Forse, J. R. Long, Science369 (2020) 392-396. doi: 10.1126/science.abb3976
C.Q. Zhu, S.H. Li, M. Luo, et al., Nat. Chem. 5 (2013) 698-703. doi: 10.1038/nchem.1690
D.F. Chen, Y.H. Hua, H.P. Xia, Chem. Rev. 120 (2020) 12994-13086. doi: 10.1021/acs.chemrev.0c00392
I. Fernández, F.M. Bickelhaupt, Chem. Soc. Rev. 43 (2014) 4953-4967. doi: 10.1039/C4CS00055B
J.J. Cabrera-Trujillo, I. Fernández, Chem. Commun. 55 (2019) 675-678. doi: 10.1039/c8cc09777a
D.L. Zhuang, A.M. Rouf, Y.Y. Li, C. Dai, J. Zhu, Chem. Asian J. 15 (2020) 266-272. doi: 10.1002/asia.201901415
M. Rosenberg, C. Dahlstrand, K. Kilså, H. Ottosson, Chem. Rev. 114 (2014) 5379-5425. doi: 10.1021/cr300471v
Y.M. Sung, M.C. Yoon, J.M. Lim, et al., Nat. Chem. 7 (2015) 418-422. doi: 10.1038/nchem.2233
B. Oruganti, P.P. Kalapos, V. Bhargav, G. London, D. Bo, J. Am. Chem. Soc. 142 (2020) 13941-13953. doi: 10.1021/jacs.0c06327
M.C. Yue, Y. Hoshino, Y. Miura, Chem. Sci. 6 (2015) 6112-6123. doi: 10.1039/C5SC01978H
P.D.L. Werz, J. Kainz, B. Rieger, Macromolecules48 (2015) 6433-6439. doi: 10.1021/acs.macromol.5b01367
X.F. Wang, H.J. Qiu, Q.S. Wu, et al., ACS Macro Lett. 9 (2020) 1611-1616. doi: 10.1021/acsmacrolett.0c00617
F. Lu, X.Z. Lin, Q.S. Wu, et al., ACS Macro Lett. 9 (2020) 266-271. doi: 10.1021/acsmacrolett.9b01031
Z.Y. Lu, J.X. Chen, H.P. Xia, Chin. J. Org. Chem. 37 (2017) 1181-1188. doi: 10.6023/cjoc201703003
M. Yan, Y. Kawamata, P.S. Baran, Chem. Rev. 117 (2017) 13230-13319. doi: 10.1021/acs.chemrev.7b00397
S.T. Farrell, C.B. Breslin, Electrochim. Acta49 (2004) 4497-4503. doi: 10.1016/j.electacta.2004.05.007
Q.J. Chen, R. Ranaweera, L. Luo, J. Phys. Chem. C122 (2018) 15421-15426. doi: 10.1021/acs.jpcc.8b03770
C.D. Jones, L.A. Lyon, Langmuir19 (2003) 4544-4547. doi: 10.1021/la034392+
R. Pelton, Adv. Colloid Interface Sci. 85 (2000) 1-33. doi: 10.1016/S0001-8686(99)00023-8
C. Wu, J. Polym. Sci. Part B: Polym. Phys. 32 (1994) 1503-1509. doi: 10.1002/polb.1994.090320822
S. Romer, F. Scheffold, P. Schurtenberger, Phys. Rev. Lett. 85 (2000) 4980-4983. doi: 10.1103/PhysRevLett.85.4980
B. Chu, In Laser Light Scattering, 2nd ed., Academic Press: New York, 1991.
Q. J. Chen, H. Zhao, T. Ming, J. F. Wang, C. Wu, J. Am. Chem. Soc. 131 (2009) 16650-16651. doi: 10.1021/ja907898u
H. Lischka, D. Nachtigallová, A. J. A. Aquino, et al., Chem. Rev. 118 (2018) 7293-7361. doi: 10.1021/acs.chemrev.8b00244
H.S. Jung, P. Verwilst, A. Sharma, et al., Chem. Soc. Rev. 47 (2018) 2280-2297. doi: 10.1039/C7CS00522A
G. McKay, Processes Impacts22 (2020) 1139-1165. doi: 10.1039/d0em00056f
K.N. Solovyov, E.A. Borisevich, Phys. Usp. 48 (2005) 231-253. doi: 10.1070/PU2005v048n03ABEH001761
D.D. Chen, D.W. Szczepanik, J. Zhu, M. Solà, Chem. Eur. J. 26 (2020) 12964-12971. doi: 10.1002/chem.202001830
O.L.I. Brown, J. Chem. Educ. 28 (1951) 428. doi: 10.1021/ed028p428
C.A. Trickett, A. Helal, B.A. Al-Maythalony, et al., Nat. Rev. Mater. 2 (2017) 1-16.
P.M. Bhatt, Y. Belmabkhout, A. Cadiau, et al., J. Am. Chem. Soc. 138 (2016) 9301-9307. doi: 10.1021/jacs.6b05345

Figure 1 (a, b) TEM images of the microgels. (c–f) Typical EDX elemental mapping of (c) carbon, (d) oxygen, (e) phosphorus, and (f) osmium on the single gel particle shown in (b).

Figure 2 (a, b) Turbidity and (c, d) DLS analysis results. (a) The δ700 and (c) < Rh>0 after CO2/N2 purge, as a function of temperature in dark. (b) A change in the δ700 and (d) < Rh>0 upon repeated 30 min of CO2/N2 purge, in dark or irradiating with an ultraviolet lamp.

Figure 3 (a) CO2 uptake capacity of the microgels under 1 atm CO2 pressure at different temperatures, and the corresponding CO2 release capacity upon N2 purge at the same temperature for the uptake. Spontaneous capture from the solution without microgels was subtracted. (b) The apparent isosteric heat of CO2 uptake (Qapp) at 20.0 ℃.

Figure 4 (a) CO2 capacity of uptake in dark upon CO2 purge, and that release in light upon CO2 purge or N2 purge. (b) Time-domain volume of CO2 in the gas flow, upon purge with 1 vol% CO2 gas. (c) CO2 uptake capacity in dark and in light, upon purge with CO2/N2 mixed-gases, and (d) changes in the < Rh>0 after the corresponding treatments. Spontaneous uptake from the solution without any microgels was subtracted. All tests were under ambient pressure at 20.0 ℃.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:
 下载:
下载: