
Figure 1.
Design of thioacetamide (TA)- and sulfonylacetamide (SA)-DABPs by the FBDD strategy.
Fragment-based discovery of sulfur-containing diarylbenzopyrimidines as novel nonnucleoside reverse transcriptase inhibitors
Sheng Han , Yuan Lei , Christophe Pannecouque , Erik De Clercq , Chunlin Zhuang , Fener Chen

Acquired immune deficiency syndrome (AIDS) that has not been eradicated is continued to present a devastating globally health problem. Etiologically, human immunodeficiency virus (HIV) is a main causal agent of AIDS [1]. According to the 2018- WHO report, 37.9 million people were infected with HIV worldwide, 1.7 million people were newly infected, and 0.77 million people died. However, only about 62% of people (23.3 million) living with HIV could receive antiretroviral therapy [2].
Nonnucleoside reverse transcriptase inhibitors (NNRTIs) are important components for the HIV highly active antiretroviral therapy (HAART), leading to remarkable reduction in AIDS-related mortality [3-6]. To date, there are six NNRTIs (Nevirapine, NVP; delavirdine, DLV; efavirenz, EFV; etravirine, ETR; rilpivirine, RPV; and doravirine, DOR) approved by FDA for HIV treatment [7]. However, not all the patients could have response to the therapy and the effectiveness of NNRTIs has been significantly reduced due to the low barrier to genetic resistance [8-10]. For instance, the K103N was one of the most prevalent NNRTI resistance-associated mutations (RAMs) for antiretroviral treatments [11-14]. The early generation NNRTIs (e.g., NVP, DLV and EFV) were reported to be ineffective against the K103N mutant [15-18]. The second generation NNRTIs, FDA-approved ETR and RPV with the diarylpyrimidine (DAPY) scaffold, showed better antiviral activity, especially against HIV mutants, potentially due to a flexible moiety of the DAPYs. However, they presented the high cytotoxicity (CC50 < 5 μmol/L) and many serious side effects have been observed, such as skin rash, liver-related, and neuropsychiatric disorders [5, 19, 20]. Moreover, the absolute oral bioavailability of ETR in human is still unknown due to its bad solubility and RPV is hardly dissolved in water (20 ng/mL at pH 7.0) [4]. The newly approved drug DOR may affect up to 1 in 10 people nausea and headache [21]. More possible side effects of DOR still need further clinical investigations. Thus, exploring new drugs with high potency and safety profile is still in an urgent demand [22-24].
On the basis of the chemical structure of DAPYs, they can be divided into three fragment parts (Fig. 1). They are two diaryl "wings" (Fragment A and Fragment B) to control the flexibility of the compounds and a core pyrimidine (Fragment C). In our previous work, we developed a series of dichloridated diarylbenzopyrimidines (DABPs) focusing on the core Fragment C and evaluating the SAR of the Fragment A [25]. The DABP 3 displayed promising EC50 values of 0.0053 μmmol/L against HIV-1 wide type (WT) and 0.0063 μmol/L against the K103N mutant strain. However, the cytotoxicity (CC50 = 9.92 μmol/L) and selectivity index were not satisfactory (SIWT = 1889, SIK103N = 1579). Moreover, the cLogP of 3 was too high (> 8, Table 1) indicating a non-drug-like property [26].
 DownLoad:
CSV
DownLoad:
CSV
 |
The impact of sulfur-containing was instrumental to the drug development [27]. Thioacetamide and sulfonyl fragments are widely used in several HIV NNRTIs, such as VRX-480773, RDEA806 and delavirdine. This strategy has been tried in our previous compound 4 [28, 29], showing promising activity and especially lower cLogP value (4.860). In this study, we hypothesized that introducing the thioacetamide fragment of compound 4 to the lead compound 3 and further oxidation on the sulfur might be favorable to the drug-like property and the antiviral activity.
The general synthetic route of compounds 5a-k and 6a-e is shown in Scheme 1. Detailed procedures and compound characterizations can be found in Supporting information. Briefly, the starting compound 7 was synthesized according to our previous work [25, 29]. Compound 7 was reacted with the various substituted α-bromoacetamides in a base condition to give TADABPs 5a-k. Then, oxidation on the sulfur by m-CPBA gave corresponding SA-DABPs 6a-e.

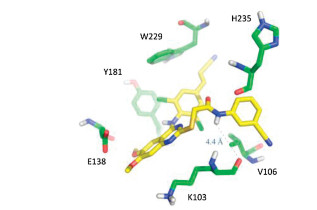
At first, we synthesized a series of thioacetamide based on 4-aminobenzonitrile substituted DABP. They decreased the cLogP compared to that of 3, although the antiviral activity was dramatically reduced in more than 200-fold. Among compounds 5a-c with cyano group, compound 5b with 3-CN showed the best antiviral activity (EC50 = 1.36 μmol/L) compared to compound 5c with 2-CN and compound 5a with 4-CN. Then, we synthesized a series of TA-DABPs 5d-k in the next round, with substitutions at meta- and ortho-positions. With electron-donating group methoxy group introduced, the cLogP values of 5d and 5e were decreased to 6.9 - 7.5. However, the antiviral activity was still unsatisfactory. Compounds 5f-k with electron withdrawing group were synthesized. They were inactive toward viral strains. Compounds 5f and 5g exhibited relatively high cytotoxicity. Molecular docking study revealed that the distance between the amide of TA-DABP 5b and the carbonyl group of K103 of the reverse transcriptase (RT) was 4.4 Å, indicating a weak interaction instead of a hydrogen-bonding interaction (Fig. 2), potentially interpreting the weak antiviral activity.
In the next round, the most potent thio-compound 5b was selected to be oxidized to SA-DABP 6a. To our delight, compound 6a (Table 2) showed better activity against the K103N mutant virus than NVP (EC50 ≥ 11.2 μmol/L) and EFV (EC50 = 0.124 μmol/L). Moreover, the cytotoxicity was decreased (CC50 = 29.8 μmol/L). In addition, compound 6a displayed an improved lipid-water partition coefficient and the cLogP was decreased to 6.6. Encouraged by the results, we synthesized and evaluated a series of SA-DABPs (6b-e) with 3-substitutions except 3-fluoro (5f) with a high cytotoxicity. Compound 6b with 3-OCH3 (EC50 = 5.12 μmol/L) showed decreased antiviral activity than 6a, but still exhibited significantly improved antiviral activity than its corresponding TA-DABP 5d (EC50 > 210 μmol/L). Compound 6c with 3-CF3 (EC50 = 1.08 μmol/L) and compound 6d (EC50 = 0.208 μmol/L) with 3-NO2 exhibited better activity against the K103N mutant virus than an early generation NNRTI NVP (EC50 ≥11.2 μmol/L). Finally, we tried to change the nitro group from meta-position to para-position to obtain compound 6e. Surprisingly, the activity of compound 6e was significantly improved while the cytotoxicity was remarkable decreased. Compound 6e (EC50 = 0.0228 μmol/L) showed ~490-fold activity than NVP (EC50 ≥ 11.2 μmol/L), ~5-fold than EFV (EC50 = 0.124 μmol/L) against the K103N mutant virus, but 10-fold lower than ETR (EC50 = 0.0037 μmol/L). Consistent with antiviral activity, compound 6e showed an IC50 of 0.98 μmol/L against the RT enzyme (Table 3), which was lower than those of 3 (IC50 = 0.046 μmol/L) and the FDA drugs. However, the CC50 value of 6e (99.6 μmol/L) was much better than them (NVP 15.0 μmol/L, EFV 6.34 μmol/L, ETR 4.59 μmol/L and RPV 4.38 μmol/L), indicating a remarkable decreased cytotoxicity. Moreover, the cLogP of 6e was decreased from 8.025 to 6.900 compared to the lead compound DABP 3. It suggested that 6e was more hydrophilic than 3 and a better solubility. Experimentally, the water solubility of 6e was 300 ng/mL at pH 7, which was higher than RPV (20 ng/mL [4]) and 3 (undetectable at 20 ng/mL).
DownLoad:
CSV
 |
To better understand the activity, we predicted the binding modes of the compounds with RT by molecular docking and dynamics (Supporting information). Comparing to 5b, the ligand torsions of 6a were changed due to the steric hindrance of the newly formed sulfonyl group, making the amide to form a new hydrogen bond with H235 (Fig. 3A). The carbonyl group of 6a was also pointed to the amide of K103 was 3.8 Å. The activity at the enzyme level also supported our prediction. Compound 6a had ~4-fold improvement in the anti-RT activity than 5b (Table 3). In Fig. 3B, the hydrogen of the amide and the nitro group of 6d could form two hydrogen bonds with K103 and V106, respectively. In Fig. 3C, two hydrogen bonds (between the amide and K103, and the amide and H235) anchored the position of compound 6e and made the binding more stable than 6d. Thus, compound 6e exhibited the 7-fold higher potency (IC50 = 0.98 μmol/L) against HIV-1 RT than 6d (IC50 = 7.01 μmol/L). In the K103N RT, besides the two hydrogen bonds similar as WT RT, the distance between the carbonyl group of 6e and the mutant amide group was 3.5 Å, indicating potential additional interactions (Fig. 3D).

In conclusion, we synthesized and evaluated two series of sulfur-containing DABPs (TA-DABPs and SA-DABPs) based on the FBDD strategy. The most potent compound 6e displayed better antiviral activity against the HIV-1 WT and K103N mutant virus than the early generation NNRTIs, and comparable activity to the second generation NNRTIs. The cytotoxicity and cLogP was significantly decreased. Molecular modelling further explained the activity and gave more guidance for further optimizations. Thus, the new sulfur-containing DABPs represent new lead compounds for anti-HIV agents, especially the HIV-1 K103N mutants.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This research was financially supported by the National Natural Science Foundation of China (No. 21372050), National Key R & D Program of China (No. 2017YFA0506000) and the Young Elite Scientists Sponsorship Program by the China Association for Science and Technology (No. 2017QNRC061). We thank Ningxia Medical University for providing the sources of molecular modeling. The technical assistance of Mr. Kris Uyttersprot, Mrs. Kristien Erven, and Mrs. Cindy Heens for the HIV experiments and HIV RT polymerase assays is gratefully acknowledged.
Supplementarymaterial related to this article canbefound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.11.020.
R.C. Gallo, L. Montagnier, N. Engl. J. Med. 349(2003) 2283-2285. doi: 10.1056/NEJMp038194
WHO website, HIV/AIDS Fact Sheet (Last Accessed August 26, 2019), https://www.who.int/hiv/data/en/.
E. De Clercq, J. Med. Chem. 62(2019) 7322-7339. doi: 10.1021/acs.jmedchem.9b00175
B. Huang, W. Chen, T. Zhao, et al., J. Med. Chem. 62(2019) 2083-2098. doi: 10.1021/acs.jmedchem.8b01729
D. Kang, H. Zhang, Z. Wang, et al., J. Med. Chem. 62(2019) 1484-1501. doi: 10.1021/acs.jmedchem.8b01656
Z. Zhou, T. Liu, D. Kang, et al., Org. Biomol. Chem. 16(2018) 1014-1028. doi: 10.1039/C7OB02828H
V. Namasivayam, M. Vanangamudi, V.G. Kramer, et al., J. Med. Chem. 62(2019) 4851-4883. doi: 10.1021/acs.jmedchem.8b00843
Y. Hsiou, J. Ding, K. Das, et al., J. Mol. Biol. 309(2001) 437-445. doi: 10.1006/jmbi.2001.4648
K. Das, E. Arnold, Curr. Opin. Virol. 3(2013) 111-118. doi: 10.1016/j.coviro.2013.03.012
K. Das, E. Arnold, Curr. Opin. Virol. 3(2013) 119-128. doi: 10.1016/j.coviro.2013.03.014
C.J. Cohen, J. Andrade-Villanueva, B. Clotet, et al., Lancet 378(2011) 229-237. doi: 10.1016/S0140-6736(11)60983-5
J. Lindberg, S. Sigurdsson, S. Lowgren, et al., Eur. J. Biochem. 269(2002) 1670-1677. doi: 10.1046/j.1432-1327.2002.02811.x
J. Ren, J. Milton, K.L. Weaver, et al., Structure 8(2000) 1089-1094. doi: 10.1016/S0969-2126(00)00513-X
Y. Yang, D. Kang, L.A. Nguyen, et al., Steitz, Elife 7(2018)E36340.
M.B.Nawrozkij, M.Forgione, A.S.Yablokov, et al., J.Med.Chem.62(2019)604-621. doi: 10.1021/acs.jmedchem.8b01238
M.T. Lai, V. Munshi, M. Lu, et al., Viruses 8(2016) E263. doi: 10.3390/v8100263
M. Udier-Blagovi c, J. Tirado-Rives, W.L. Jorgensen, et al., J. Am. Chem. Soc. 125(2003) 6016-6017. doi: 10.1021/ja034308c
K. Das, A.D. Clark Jr., P.J. Lewi, et al., J. Med. Chem. 47(2004) 2550-2560. doi: 10.1021/jm030558s
J. Adams, N. Patel, N. Mankaryous, M. Tadros, C.D. Miller, Ann. Pharmacother. 44(2010) 157-165. doi: 10.1345/aph.1M359
A.Blas-Garcia, J.V.Esplugues, N.Apostolova, Curr.Med.Chem.18(2011)2186-2195. doi: 10.2174/092986711795656180
European Medicines Agency web site, Pifeltro: European Public Assessment Report-Medicine Overview, 2019. (Last Accessed August 26, 2019) https://www.ema.europa.eu/en/medicines/human/EPAR/pifeltro
X. Li, B. Chen, L. Lan, et al., Chin. Chem. Lett. 29(2018) 1637-1640. doi: 10.1016/j.cclet.2018.06.003
Y. Guo, L. Fu, X. Fan, X. Shi, Chin. Chem. Lett. 29(2018) 1167-1170. doi: 10.1016/j.cclet.2018.03.024
W.H. Zhou, X.G. Xu, J. Li, et al., Chin. Chem. Lett. 28(2017) 422-425. doi: 10.1016/j.cclet.2016.09.001
S. Han, Y. Sang, Y. Wu, et al., ACS Infect. Dis. (2019), doi: http://dx.doi.org/10.1021/acsinfecdis.9b00229.
M.D. Shultz, J. Med. Chem. 62(2019) 1701-1714. doi: 10.1021/acs.jmedchem.8b00686
E.A. Ilardi, E. Vitaku, J.T. Njardarson, J. Med. Chem. 57(2014) 2832-2842. doi: 10.1021/jm401375q
Z.Y. Wan, J. Yao, T.Q. Mao, et al., Eur. J. Med. Chem. 102(2015) 215-222. doi: 10.1016/j.ejmech.2015.08.007
Z.Y. Wan, Y. Tao, Y.F. Wang, et al., Bioorg. Med. Chem. 23(2015) 4248-4255. doi: 10.1016/j.bmc.2015.06.048

Figure 1 Design of thioacetamide (TA)- and sulfonylacetamide (SA)-DABPs by the FBDD strategy.

Scheme 1 Synthesis route of compounds 5a-k and 6a-e. Reagents and conditions: (a) substituted α-bromoacetamide, t-BuOK, DMF, r.t., 1 h; (b) m-CPBA, CH2Cl2/DMF, -78 ℃ - r.t.

Figure 2 Predicted binding modes of compound 5b (carbons in yellow) with the HIV-1 RT (PDB: 6C0N). Residues involved in interactions are shown as green sticks.

Figure 3 Predicted binding modes of compounds (carbons in yellow) with the HIV-1 WT and K103N mutant RT (PDB: 6C0N). (A) RT with 6a; (B) RT with 6d; (C) RT with 6e; (D) K103N mutant RT with 6e. Residues involved in interactions are shown as green sticks. Mutated residues are depicted as cyan sticks. The distances are depicted as black dashed lines.
Table 1. Activity of 5a-l against HIV-1 wide type (ⅢB strain), K103N mutant strain and cytotoxicity in MT-4 cells.a
| |
 下载: 导出CSV
下载: 导出CSV
Table 2. Activity of 6a-f against HIV-1 ⅢB, K103N strains and cytotoxicity in MT-4 cells.a
| |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们

 下载:
下载: