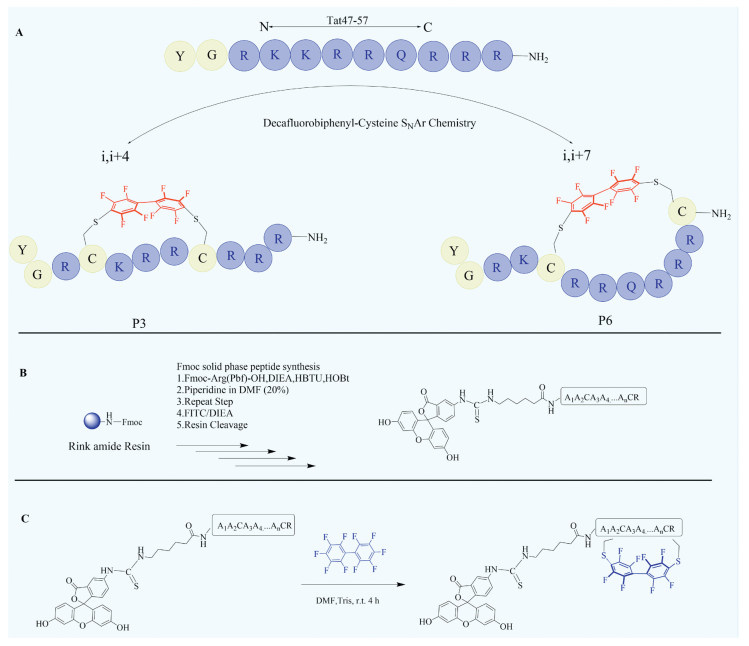
Scheme 1.
Tat47-57 analogues design by decafluorobiphenyl-based peptides staple (A), conventional Fmoc-solid-phase peptide synthesis (SPPS, B) and decafluorobiphenyl-based stapled peptides synthesis (C).
Achieving enhancing endosome escape of Tat47-57-derived stapled peptides through decafluorobiphenyl bridge
Shu Li , Yuanyuan Tang , Xiaojing Liu , Shibo Song , Baokang Zhu , Min Chang , Yali Peng

Cell membrane, consisting of a lipid bilayer, membrane-embedded proteins, carbohydrates and their conjugates, is a barrier that controls material transportation [1,2]. Many biomacromolecules such as proteins, peptides, and nucleic acids possess highly potent and specific biological activities in vitro but cannot be used as therapeutic agents or research tools because they often show poor cell permeability. A series of strategies such as viral vectors, nanomaterial-based delivery systems and direct injection, are developed to overcome this hurdle, but the efficiency and safer are still need to be improved [3-8]. Since the highly positively charged HIV Tat peptide, 47YGRKKRRQRRR57 was discovered, one potential strategy for cytosolic delivery based on "cell-penetrating peptides" (CPPs) has been developed in biomedical research [9,10]. CPPs have been used to deliver small-molecule drugs, peptides, proteins, and nucleic acids into cultured mammalian cells and live organisms [11-13]. Generally, CPPs deliver the biologics into mammalian cells through endocytosis pathways, which need a highly endosomal escape efficiency to avoid degradation in lysosomes [14-17]. Unfortunately, the endosomal membrane has proven to be a significant barrier toward cytoplasmic delivery by these CPPs. Therefore, the development of highly escape efficient CPPs becomes a desirable solution. Due to the close similarity of the cell membrane and endosome membrane, it is difficult to design peptides that can recognize specific lipids. In recent years, new approaches toward effective CPPs have been investigated, particularly, the strategy of designing peptides that can increase their endosomal escape efficiency. For instance, a family of small amphipathic cyclic peptides was showed to be highly efficient CPPs, with highly cytosolic delivery efficiencies [18-23]. The hydrocarbon stapled Tat47-57 analogues with α-helix exhibited high endosomal escape efficiency [7]. The cell-penetrating peptide-bismuth bicycles exhibit efficient cellular uptake at concentrations as low as 10 nmol/L [22]. Despite these significant advances, the design and discovery of highly efficient CPPs call for further exploration.
Fluorine chemistry has been developed for more than 200 years, and fluorinated organic compounds are widely applied in a significant area, such as pharmaceuticals, agrochemicals, materials [24-27]. Unique properties emerge when fluorine atom is introduced into molecules due to its largest electronegativity and second smallest size. The introduction of fluorine imparts various properties to molecules, such as molecular conformation, dipole moment, acidity, and alkalinity, which would affect the toxicity, selectivity, potency, and pharmacodynamic and pharmacokinetic properties [28,29]. Bradley et al. reported methodology that allows for the macrocyclization of an unprotected peptide between two cysteine residues with a perfluoroaryl linker through SNAr chemistry [30,31]. They demonstrated that these perfluoroarene-based peptide macrocycles had increased cellular uptake and improved the ability of peptides to cross the blood−brain barrier compared with their linear counterparts [32,33]. However, this paper represents the first application of the DFBP strategy to investigate enhanced endosomal escape. In this study, fluorine-containing stapled bridge was introduced to peptide molecules and their effect of cell penetrating activity was investigated. We discovered that decafluorobiphenyl-based stapled analogues of Tat47-57 (P3 and P6) are highly active CPPs, capable of delivering a variety of cargo molecules into the cytosol of mammalian cells. We also found that the decafluorobiphenyl staple plays a key role in cellular uptake and endosomal escape. Two peptides, P3 and P6 exhibited prominent cell permeability, presumably clathrin-dependent endocytosis. They also showed endosomal escaping ability, and applying P3 and P6 as CPPs for delivering phosphopeptides (sequence: GpYEEI) and avidin has been accomplished.
We designed decafluorobiphenyl-based stapled peptides based on the previously reported natural short-chain cell-penetrating peptide Tat47-57, which is derived from the transactivating transcriptional activator of human immunodeficiency virus 1 [9]. A series of decafluorobiphenyl-based stapled analogues of Tat47-57 were synthesized using conventional Fmoc-solid-phase peptide synthesis (SPPS), followed by decafluorobiphenyl-cysteine SNAr chemistry (Scheme 1). In this library, analogues of this model peptide, Tat47-57, were prepared containing two cysteine residues in a variety of i, i + 4 or i, i + 7 configurations (Table 1). Cysteines were substituted rather than inserted into the sequence to maintain the spatial relationships between the other amino acids of the peptide, except P6. Five parameters of the decafluorobiphenyl-based staples were systematically varied in the library: (1) Position of the decafluorobiphenyl-based staple (P1-P6); (2) Biphenyl bridge, non-containing fluorine (P3b and P6b); (3) Hexafluorobenzene bridge, decreasing fluorine and benzene number (P3a); (4) Disulfide bridge (P3c and P6c); (5) Modified with decafluorobiphenyl without cyclization (P3d, P3e, P6d and P6e). We manipulated the fluorine groups of staple bridge by substituting decafluorobiphenyl with 4,4′-bis(bromomethyl)biphenyl. With variations in the staple position and type, a panel of 17 conformationally constrained analogues was constructed. All the peptides were purified by reversed-phase high performance liquid chromatography (RP-HPLC) and were found to have > 95% purity (Table 1, Table S1 and Fig. S8 in Supporting information).
 DownLoad:
CSV
DownLoad:
CSV
| Entry | Name | Sequence | Position | Purity (%) | Theoretical MW | Measured MW |
| 1 | Tat | FITC-Acp-YGRKKRRQRRR-NH2 | None | 99.17 | 2060.09 | 2061.11 |
| 2 | P1 |  |
1+8 | 99.95 | 2268.95 | 2270.56 |
| 3 | P2 |  |
2+9 | 98.43 | 2346.95 | 2348.56 |
| 4 | P3 |  |
4+8 | 99.96 | 2303.92 | 2305.61 |
| 5 | P4 |  |
4+11 | 99.94 | 2275.88 | 2277.48 |
| 6 | P5 |  |
5+9 | 99.67 | 2275.88 | 2277.48 |
| 7 | P6 |  |
5+12 | 99.96 | 2431.98 | 2432.95 |
| 8 | uP3 | FITC-Acp-YGRCKRRCRRR-NH2 | None | 95.43 | 2009.95 | 2010.96 |
| 9 | P3a |  |
4+8 | 99.84 | 2155.93 | 2156.91 |
| 10 | P3b |  |
4+8 | 99.48 | 2188.03 | 2189.04 |
| 11 | P3c |  |
4+8 | 99.69 | 2007.94 | 2008.95 |
| 12 | P3d |  |
None | 99.01 | 2348.98 | 2349.94 |
| 13 | P3e |  |
None | 96.47 | 2349.01 | 2349.97 |
| 14 | uP6 | FITC-Acp-YGRKCRRQRRRCRR-NH2 | None | 97.90 | 2431.98 | 2432.95 |
| 15 | P6b |  |
5+12 | 99.48 | 2316.09 | 2317.09 |
| 16 | P6c |  |
5+12 | 99.48 | 2138.01 | 2139.02 |
| 17 | P6d |  |
None | 99.45 | 2348.98 | 2349.95 |
| 18 | P6e |  |
None | 96.90 | 2477.07 | 2478.03 |
 | ||||||
Circular dichroism (CD) spectroscopy was performed to measure the secondary structures of our novel designed peptides. Previous studies demonstrated that varying the environment affected the biological activity of cell penetrating peptides by changing their conformation. The secondary structures of the new stapled analogues in water membrane-mimetic environment (50% trifluoroethanol [TFE]) were measured to explore the relationship between the conformation and the cellular uptake of the new stapled analogues. The CD data exhibited that neither the parent peptide Tat47-57 nor the stapled analogues displayed a specific secondary structure in water or TFE/H2O (1:1, v/v, Fig. S1 in Supporting information). These results suggested that decafluorobiphenyl-based staple might not induce formation of the secondary structures of the new peptides. Taking both the secondary structures and the related cellular uptake into consideration, it was found that a well-defined secondary structure is not essential for the newly stapled analogues to achieve a satisfactory cellular uptake or endosome escape.
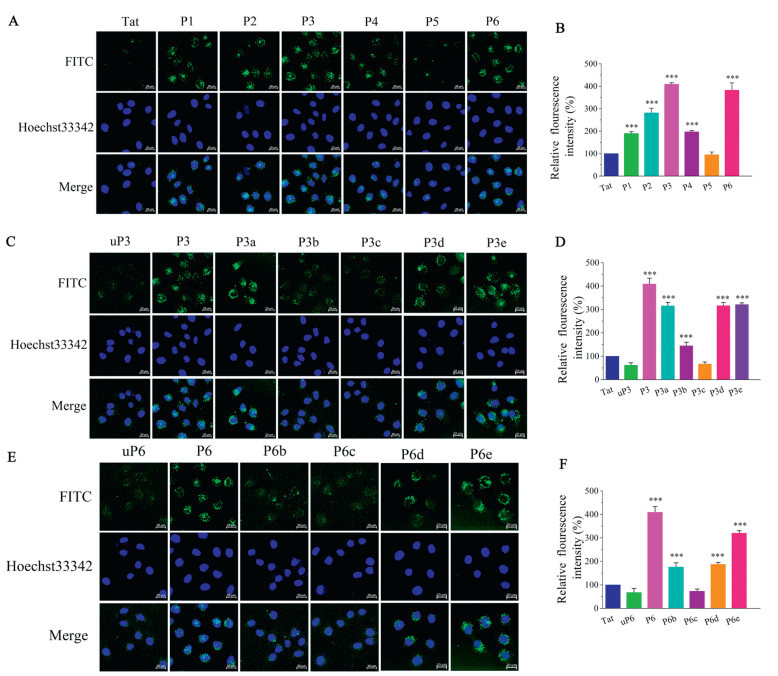
In order to visualize and quantify the cell-penetrating efficiency of these new designed stapled analogues, we labeled their N-termini with fluorescein isothiocyanate (FITC). To avoid techniques that require the fixation of cells as fixation tends to generate false-positive results, a combination of fluorescence confocal microscopy and flow cytometry, both employing live cells were carried to detect cell-penetrating abilities of our novel designed stapled peptides. Flow cytometry showed that the mean fluorescence intensity (MFI) of analyzed cells reveals, for all analogues, except for P5, a higher uptake than for the positive control group using Tat47-57 (Fig. 1). Among all analogues, P3 and P6 conveyed in highest fluorescence intensities at efficiencies 3.5−4-fold higher than that of Tat47-57, while P5 showed lower uptake (Fig. 1B). Using confocal microscopy, we confirmed these effects of analogues on HeLa (Fig. 1A). To confirm that the fluorescence observed in flow cytometry is intracellular and not associated with cell surface, uptake experiments of Tat47-57, P3 and P9 were repeated in the presence of Trypan Blue to quench extracellular fluorescence (Fig. S2 in Supporting information). No major loss of fluorescence was observed in the presence of Trypan Blue in the case of P3 and P6, indicating that the observed fluorescence is indeed intracellular. Moreover, to assess the impact of fluorine, biphenyl linker and decafluorobiphenyl bridge, the cellular uptake of uP3, P3a, P3b, P3c, P3d, P3e, uP6, P6b, P6c, P6d and P6e was measured using flow cytometry. Interestingly, when replaced a decafluorobiphenyl bridge with a biphenyl bridge (P3 vs. P3b, P6 vs. P6b), MFI of P3b and P6b had significantly decreased (Figs. 1D and F), demonstrating that the introduction of F atoms into peptides is key to CPPs activities. P3a was stapled with a hexafluorobenzene bridge. Its cellular uptake was significantly lower than that of P3, illustrating that F atoms and the biphenyl bridge both greatly impact the CPPs activity. P3c and P6c with a disulfide bridge, were less active than their model peptide Tat47-57, illustrating aromatic bridge and F atoms impart their cell permeability. In addition, the non-stapled peptides (uP3 and uP6) were less active than their stapled peptides counterparts (P3 and P6). Interestingly, P3d, P3e, P6d and P6e, modified with decafluorobiphenyl without cyclization, showed excellent cellular uptake, but it is lower than that of P3 and P6. These results indicate that both decafluorobiphenyl and staple play an important role in cellular uptake. Confocal microscopy imaging subsequently confirmed these effects on HeLa cells (Figs. 1C and E). These results clearly demonstrated that F atoms and the aromatic bridge staple are important factors that determine the cell penetrating efficiency. Negatively charged cell-surface glycans have been indicated to interact with cationic cell penetrating peptides and have an influence on the uptake of cell penetrating peptides [34,35]. As shown in Fig. S3 (Supporting information), it was found that cellular uptake of the analogues was related to their binding with heparin (a close analogue of heparan sulfate). Notably, for P4 and P5, cellular uptake was not affected by cell-surface heparan sulfate, suggesting that there may be other negatively charged cell-surface glycans. We therefore determined the binding affinity of the examined cell penetrating peptides to heparin by Isothermal Titration Calorimetry (Table S2 and Fig. S4 in Supporting information). These experiments revealed comparable binding affinities of heparin to P3 (Kd = 0.054 ± 0.005 µmol/L) and P6 (Kd = 0.041 ± 0.005 µmol/L). Heparin binds to P3 and P6 3–4-fold tighter compared to Tat47-57 (Kd = 0.160 ± 0.015 µmol/L). These binding affinities of the CPPs to heparin correlate well with their uptake efficiency.
To investigate the endosomal membranolytic behavior of the lead template peptide Tat47-57, as well as decafluorobiphenyl stapled analogues, lipid vesicles were employed to simulate endosomal membranes for the assessment of calcein dye leakage. Calcein dye was encapsulated in large unilamellar vesicles (LUVs, Table S3 in Supporting information), and calcein dye release was examined to evaluate the disruption of lipid vesicles. As shown in Fig. 2, at concentrations of 5 µmol/L or 10 µmol/L, all peptides exerted a leakage effect on lipid vesicles mimicking endosomal membranes (< 10% leakage) after 30 min of incubation. Leakage effect of all analogues increased in a dose-dependent manner. Notably, Tat47-57 showed a lowest leakage on LUVs, while decafluorobiphenyl stapled analogues (P1-P6) exerted a clearly increased leakage effect (Fig. 2A). In comparison with P3a, P3b, P3c and uP3, we observed that P3 has a higher leakage effect (Fig. 2B). Similarly, for P6 and its analogues, the consistent result was obtained as shown in Fig. 2C. The results of calcein dye leakage experiments suggested that decafluorobiphenyl stapled analogues have a potent effect on the disruption of the endosomal membrane. F atoms act the critical role in selectively disrupting endosomal membranes.
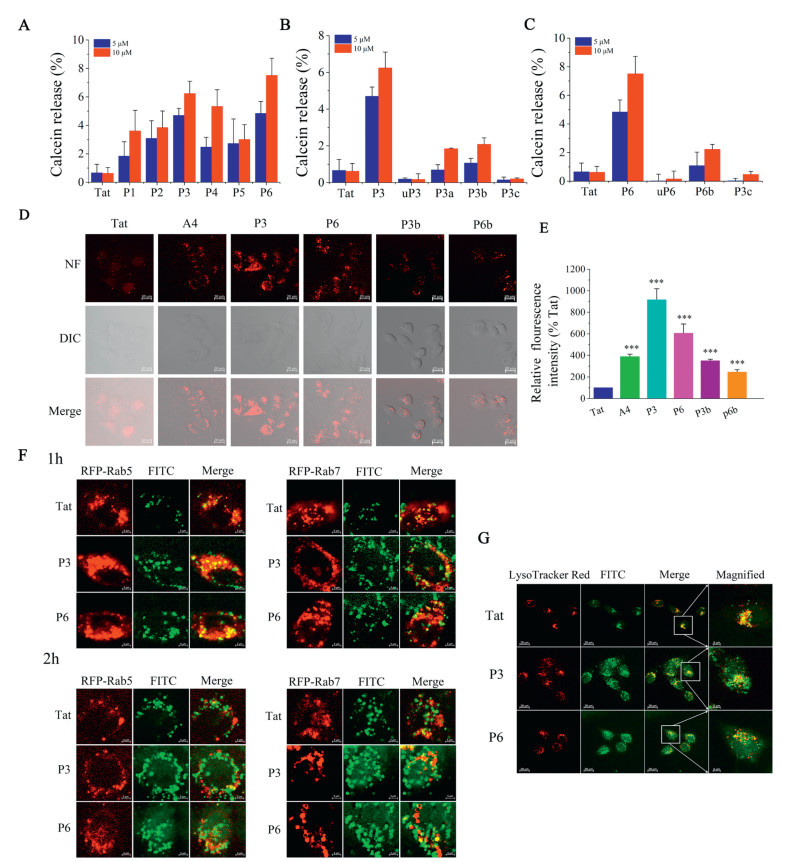
To further assess the endosomal escape behavior of our analogues, a quantitative measurement of endosomal escape was performed by naphthofluorescein-labeled CPPs, which is a pH-sensitive fluorophore and was reported to quantitate the endosomal escape of CPPs [36]. As shown in Figs. 2D and E, the results of flow cytometry showed that P3 and P6 have a potent ability of endosomal escape (9-fold and 6-fold better than Tat47-57), while P3b and P6b are slightly better than Tat47-57 (3.4-fold and 2.8-fold). In comparison to P3 and P6, the MFI of P3b and P6b had significantly decreased, suggesting that F atoms of decafluorobiphenyl bridge was essential for selectively disrupting endosomal membranes. These data were consistent with the results confirmed by confocal microscopy in live HeLa cells. Intracellular localization was achieved with the late endosomes/lysosomes counter-stain LysoTracker Red (red) in live HeLa cells via confocal microscopy. As shown in Fig. 2G, we observed that most of the green fluorescence of FITC-P3 and FITC-P6 were located in the cytoplasm, which suggesting that P3 and P6 have a better endosomal escape efficiency compared to Tat47-57. Together, these results suggest that decafluorobiphenyl stapled analogues P3 and P6 have the high cytosolic delivery efficiency.
Rab5 is a master regulator of endosome biogenesis and recruits additional cellular factors required for vesicle maintenance, fusion, and maturation [37]. The resulting early endocytic compartment mixes via homotypic fusion with other Rab5+ vesicles and delivers cargo to other cellular locales through the budding off of transport vesicles. Some cargoes are delivered to late endosomes, marked by Rab7, for degradation in lysosomes [38]. To characterize the intracellular route taken by Tat47-57, P3 and P6, we looked for overlap of these molecules with markers of endocytic uptake and RFP-tagged Rab proteins. HeLa cells were transfected with GFP-Rab5 and treated for 1 or 2 h with 5 µmol/L Tat47-57, P3 and P6 (Fig. 2F). When these cells were examined by confocal microscopy, the major FITC signal overlapped with the signal from RFP-Rab5, confirming that Tat47-57, P3 and P6 were present in Rab5+ vesicles. Because Rab5 vesicles subsequently deliver their cargo to downstream vesicles, we also evaluated colocalization of Tat47-57, P3 and P6 with RFP-Rab7. HeLa cells transfected with RFP-Rab7 and treated with Tat47-57, P3 and P6 for 2 h as above showed a little fraction of the FITC signal located in the RFP-Rab7 compartment, confirming that P3 and P6 enter early (Rab5+) and escape from late (Rab7+) endosomes or enter the lysosome (Fig. 2F). HeLa cells were treated with Tat47-57, P3 and P6 for 2 h after staining late endosomes/lysosomes with LysoTracker Red (red, Fig. 2G). Some fraction of the FITC signal located in the late endosomes/lysosomes compartment, suggesting that Tat47-57, P3 and P6 escape from late (Rab7+) endosomes.
In order to probe the possible involving mechanism of P3 and P6, we incubated HeLa cells in the presence of different inhibitors or at different temperatures to inhibit endocytosis comprehensively. The cells were treated with various inhibitors, including ethylisopropylamiloride (EIPA, a macropinocytosis inhibitor) [39], chlorpromazine (CPZ, a clathrin-mediated pathway inhibitor) [40], filipin (a caveolae-mediated endocytosis inhibitor) [41], and methyl-β-cyclodextrin (M-β-CD, a lipid raft-mediated endocytosis inhibitor) [42]. The followed cell permeability evaluation indicated that, on the one hand, the cell uptake of FITC-labeled P3 or P6 was reduced to almost the basal level at 4 ℃. On the other hand, the use of CPZ resulted in a decrease of approximately 41% and 45% in peptide uptake for P3 and P6, respectively. In contrast, the treatment of cells using EIPA, or filipin, or M-β-CD did not significantly impact the cell uptake of P3 or P6 (Fig. S6 in Supporting information). These results together indicated that the cellular uptake of P3 or P6 is energy-dependent, and likely mainly through clathrin-mediated endocytosis pathways. Cytotoxicity and hemolytic properties are key indexes to evaluate drug safety. Cytotoxicities of Tat47-57 and the newly designed stapled analogues in HeLa, NIH3T3 and CHO-K1 cells were estimated using a MTT assay, analysis of which revealed that none of the stapled analogues (P3 and P6) exhibited obvious toxicity, and their cell viability was remained above 95% even at the highest tested concentration (50 µmol/L, Fig. S7A in Supporting information). Moreover, results from the lactate dehydrogenase (LDH) release assay confirmed that no obvious disruption of the cell membrane was caused by either the P3 or P6 peptide (Fig. S7B in Supporting information). Then, to further understand the drug safety, Hemolytic toxicities of Tat47-57 and the newly designed stapled analogues were evaluated using the red blood cells of mice. The results showed that both Tat47-57 and analogues (P3 and P6) were safe to the cells with hemolysis values < 5% even at the highest tested concentration (100 µmol/L, Fig. S7C in Supporting information). Because of better cellular uptake efficiency and safety of analogues P3 and P6, they were further evaluation.
Negatively charged phosphopeptides do not readily cross cellular membranes, thus studying these peptides in cellular system is challenging. Phosphopeptides mimic the interactions between the negatively charged phosphate group of phosphoproteins and positively charged amino acids in the binding pockets of various of proteins, which are used as probes for exploring phosphoprotein–protein interactions. We evaluated the cellular delivery of negatively charged phosphopeptides using the Tat47-57, P3 and P6 peptides. The intracellular location of the phosphopeptides was monitored by using confocal microscopy. By treating the HeLa cells with a mixture of Tat47-57, P3 or P6 and phosphopeptides, both P3 and P6 were able to enhance the cellular uptake of the cargo phosphopeptides. However, Tat47-57 was hardly able to deliver phosphopeptides into the cells (Figs. 3A and B). The labeled phosphopeptides alone did not show any cellular uptake. The image confirmed significant uptake of GpYEEI in the presence of the stapled analogues. Further, the intracellular localization of the phosphopeptides/the decafluorobiphenyl stapled analogues complexes was examined with confocal microscopy after staining late endosomes/lysosomes with LysoTracker Red (red, Fig. 3C). As shown in Fig. 3C, punctate signals distribute in the cytosol and overlap with the lysosome tracker for the phosphopeptides/Tat47-57 complex. In contrast, diffused FITC signals were observed in the cells for the phosphopeptides/the decafluorobiphenyl stapled analogues complexes, indicating a possible endosomal escape of decafluorobiphenyl stapled analogues. Take the above results together, decafluorobiphenyl stapled analogues led to an increase in the intracellular delivery of negatively charged phosphopeptides.
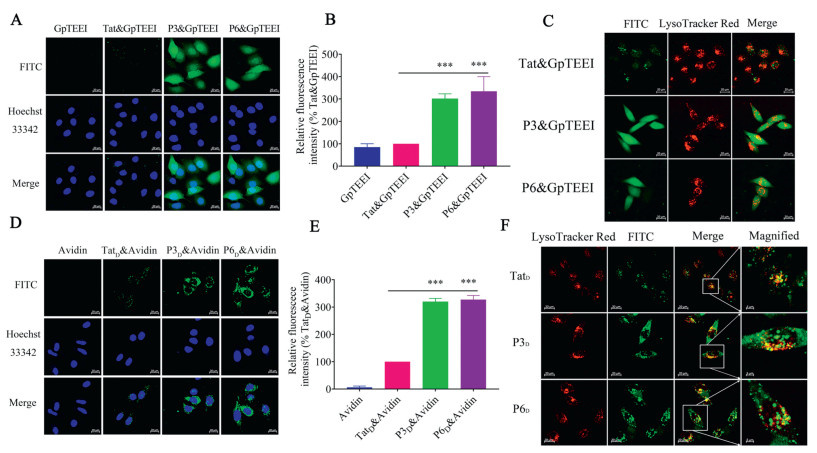
Based on the above experiments, we further evaluated the cellular delivery of biomacromolecules (avidin) utilizing the decafluorobiphenyl stapled analogues. Biotinylated P3 and P6 (Table S1 in Supporting information) were treated with FITC labeled avidin to form peptide-avidin complexes through biotin avidin interactions. The detailed methods are provided in Supporting information. Then, by treating the HeLa cells with these complexes, the result of flow cytometry showed that P3 and P6 delivered avidin to cells at 319% and 327% higher efficiencies, respectively, compared to Tat47-57 (Fig. 3E). These results were confirmed by live-cell confocal microscopy as shown in Fig. 3D. In order to confirm the endosome-escaping ability of decafluorobiphenyl stapled analogues-avidin complexes, the intracellular localization of the decafluorobiphenyl stapled analogues-avidin complex examined by confocal microscopy after staining late endosomes/lysosomes with LysoTracker Red (red, Fig. 3F). For Tat47-57-avidin complex, some punctate signals distribute in the cytosol and overlap with the lysosome tracker, a little cytosolic diffused signal. But some cytosolic diffused signals were observed when we evaluated the decafluorobiphenyl stapled analogues-avidin complexes, only little punctate signals distribute in the cytosol and overlap with the lysosome. These results suggest that decafluorobiphenyl stapled analogues exhibited potential biomacromolecules intracellular delivery ability.
In summary, a library of decafluorobiphenyl-based stapled peptides with variations in the position of stapling, the type of staples, and fluorine atoms of bridge was created. A detailed investigation of cellular uptake and endosomal escape activities revealed multiple pieces of insightful information. Among these analogs, P3 and P6 were found to possess several advantages, such as superior cellular uptake efficiency and the ability to escape from late (Rab7+) endosomes. The results of calcein dye leakage and a pH-sensitive fluorophore demonstrated considerable endosomal escape efficiency of P3 and P6. Our study also suggested that decafluorobiphenyl bridge had an outstanding effect on endosmal escape. Coupled with their high endosomal escape efficiency and ease of preparation, these stapled CPPs should provide a very useful class of general membrane transporters for cytosolic delivery of chemical probes and proteins.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Shu Li: Writing – review & editing, Project administration, Funding acquisition. Yuanyuan Tang: Formal analysis, Data curation, Conceptualization. Xiaojing Liu: Methodology, Investigation. Shibo Song: Validation, Software, Methodology. Baokang Zhu: Supervision, Software. Min Chang: Writing – review & editing, Writing – original draft, Project administration, Funding acquisition. Yali Peng: Writing – review & editing, Writing – original draft.
The study was supported by the grants from the National Natural Science Foundation of China (No. 82404426), the Natural Science Foundation of Gansu Province (No. 18JR3RA280), the Funds for Fundamental Research Creative Groups of Gansu Province (No. 20JR5RA310), Shihezi University High-level Talent Research Start-up funds (No. RCZK202446), and Tianchi Talent Introduction Plan. In addition, the authors would like to thank Core Facility of School of Life Sciences, Lanzhou University for supporting this work.
Supplementary material associated with this article can be found, in the online version, at doi:
S.B. Gould, Curr. Biol. 28 (2018) R381–R385. doi: 10.1016/j.cub.2018.01.086
E. Sezgin, I. Levental, S. Mayor, Nat. Rev. Mol. Cell Biol. 18 (2017) 361–374. doi: 10.1038/nrm.2017.16
J.J. Shi, P.W. Kantoff, R. Wooster, O.C. Farokhzad, Nat. Rev. Cancer 17 (2017) 20–37. doi: 10.1038/nrc.2016.108
W.A. Lim, C.H. June, Cell 168 (2017) 724–740. doi: 10.1016/j.cell.2017.01.016
D. Furtado, M. Björnmalm, S. Ayton, et al., Adv. Mater. 30 (2018) e1801362. doi: 10.1002/adma.201801362
M. Sadelain, I. Rivière, S. Riddell, Nature 545 (2017)423–431. doi: 10.1038/nature22395
S. Li, X.J. Zhang, C. Guo, et al., Chem. Commun 56 (2020) 15655–15658. doi: 10.1039/d0cc06312f
S.A. Khan, M.J. Akhtar, Bioorg. Chem. 120 (2022) 105599. doi: 10.1016/j.bioorg.2022.105599
A.D. Frankel, C.O. Pabo, Cell 55 (1988) 1189–1193. doi: 10.1016/0092-8674(88)90263-2
M. Green, P.M. Loewenstein, Cell 55 (1988) 1179–1188. doi: 10.1016/0092-8674(88)90262-0
J.M. Wu, S. Roesger, N. Jones, C.J. Hu, S.D. Li, J. Control. Release 366 (2024) 864–878. doi: 10.1016/j.jconrel.2024.01.038
J.Y. Lin, Z.H. Yu, X.L. Gao, ACS Nano 18 (2024) 22752–22779. doi: 10.1021/acsnano.4c06851
I. Nakase, H. Akita, K. Kogure, et al., Acc. Chem. Res. 45 (2012) 1132–1139. doi: 10.1021/ar200256e
S.H. Medina, S.E. Miller, A.I. Keim, et al., Angew. Chem. Int. Ed. 55 (2016) 3369–3372. doi: 10.1002/anie.201510518
S. Mandal, G. Mann, G. Satish, A. Brik, Angew. Chem. Int. Ed. 60 (2021) 7333–7343. doi: 10.1002/anie.202016208
A. Klipp, M. Burger, J.C. Leroux, Adv. Drug Deliv. Rev. 200 (2023) 115047. doi: 10.1016/j.addr.2023.115047
Z.Q. Qian, A. Martyna, R.L. Hard, et al., Biochemistry 55 (2016) 2601–2612. doi: 10.1021/acs.biochem.6b00226
Z.Q. Qian, T. Liu, Y.Y. Liu, et al., ACS Chem. Biol. 8 (2013) 423–431. doi: 10.1021/cb3005275
Z.Q. Qian, J.R. LaRochelle, B.S. Jiang, et al., Biochemistry 53 (2014) 4034–4046. doi: 10.1021/bi5004102
M. Oba, M. Kunitake, T. Kato, A. Ueda, M. Tanaka, Bioconjug. Chem. 28 (2017) 1801–1806. doi: 10.1021/acs.bioconjchem.7b00190
D. Mandal, A.N. Shirazi, K. Parang, Angew. Chem. Int. Ed. 50 (2011) 9633–9637. doi: 10.1002/anie.201102572
S. Voss, L.D. Adair, K. Achazi, et al., Angew. Chem. Int. Ed. 63 (2024) e202318615. doi: 10.1002/anie.202318615
A. Saha, S. Mandal, J.V.V. Arafiles, et al., Angew. Chem. Int. Ed. 61 (2022) e202207551. doi: 10.1002/anie.202207551
Y. Wang, X. Yang, Y.F. Meng, Chem. Rev. 124 (2024) 3494–3589. doi: 10.1021/acs.chemrev.3c00826
Z.H. Guo, Q. Yu, Y.C. Chen, et al., Chem. Rec. 23 (2023) e202300108. doi: 10.1002/tcr.202300108
H.B. Mei, J.L. Han, K.D. Klika, et al., Eur. J. Med. Chem. 186 (2020) 111826. doi: 10.1016/j.ejmech.2019.111826
R. Hevey, Chemistry 27 (2021) 2240–2253. doi: 10.1002/chem.202003135
R.J. Glyn, G. Pattison, J. Med. Chem. 64 (2021) 10246–10259. doi: 10.1021/acs.jmedchem.1c00668
P. Richardson, Expert. Opin. Drug Discov. 16 (2021) 1261–1286. doi: 10.1080/17460441.2021.1933427
A.M. Spokoyny, Y.K. Zou, J.J. Ling, et al., J. Am. Chem. Soc. 135 (2013) 5946–5949. doi: 10.1021/ja400119t
Y.K. Zou, A.M. Spokoyny, C. Zhang, et al., Org. Biomol. Chem. 12 (2014) 566–573. doi: 10.1039/C3OB42168F
C.M. Fadzen, J.M. Wolfe, C.F. Cho, et al., J. Am. Chem. Soc. 139 (2017) 15628–15631. doi: 10.1021/jacs.7b09790
C. Ngambenjawong, J.M. Pineda, S.H. Pun, Bioconjug. Chem. 27 (2016) 2854–2862. doi: 10.1021/acs.bioconjchem.6b00502
E.G. Stanzl, B.M. Trantow, J. R. Vargas, P.A. Wender, Acc. Chem. Res. 46 (2013) 2944–2954. doi: 10.1021/ar4000554
M. Li, S. Schlesiger, S.K. Knauer, C. Schmuck, Angew. Chem. Int. Ed. 54 (2015) 2941–2944. doi: 10.1002/anie.201410429
Z.Q. Qian, P.G. Dougherty, D. Pei, Chem. Commun. 51 (2015) 2162–2165. doi: 10.1039/C4CC09441G
H. Stenmark, R.G. Parton, O. Steele-Mortimer, et al., EMBO J. 13 (1994) 1287–1296. doi: 10.1002/j.1460-2075.1994.tb06381.x
J.S. Appelbaum, J.R. LaRochelle, B.A. Smith, et al., Chem. Biol. 19 (2012) 819–830. doi: 10.1016/j.chembiol.2012.05.022
C. Commisso, S.M. Davidson, R.G. Soydaner-Azeloglu, et al., Nature 497 (2013) 633–637. doi: 10.1038/nature12138
L.H. Wang, K.G. Rothberg, R.G. Anderson, J. Cell Biol. 123 (1993) 1107–1117. doi: 10.1083/jcb.123.5.1107
J.E. Schnitzer, P. Oh, E. Pinney, J. Allard, J. Cell Biol. 127 (1994)1217–1232. doi: 10.1083/jcb.127.5.1217
H.T. Ruan, X.S. Chen, C. Xie, et al., ACS Appl. Mater. Interfaces 9 (2017) 17745–17756. doi: 10.1021/acsami.7b03682

Scheme 1 Tat47-57 analogues design by decafluorobiphenyl-based peptides staple (A), conventional Fmoc-solid-phase peptide synthesis (SPPS, B) and decafluorobiphenyl-based stapled peptides synthesis (C).

Figure 1 Evaluation of decafluorobiphenyl-based stapled peptides for CPP candidates. Confocal micrographs of HeLa cells incubated with 5 µmol/L of FITC-conjugated P1-P6 (A), uP3-P3e (C) and uP6-P6e (E) for 2 h. Scale bar = 20 µm. Uptake of P1-P6 (B), uP3-P3e (D) and uP6-P6e (F) by flow cytometry (FACS). HeLa cells were incubated with 5 µmol/L of peptides for 2 h. All results are the mean ± SEM (n = 4). ***P < 0.001, 0.001 ≤ **P < 0.01, 0.01 ≤ *P < 0.05 vs. Tat.

Figure 2 Endosomal escape efficiency of decafluorobiphenyl-based stapled peptides. Leakage of calcein dye from endosomal membrane mimicking liposomes (A-C) in PBS buffer at pH 5.5. Results are the mean ± SEM (n = 4). (D) Confocal micrographs of HeLa cells incubated with 5 µmol/L of NF-conjugated peptides for 2 h. Scale bar = 20 µm. (E) Uptake of these peptides by flow cytometry (FACS). HeLa cells were incubated with 5 µmol/L of NF-conjugated peptides for 2 h. Results are the mean ± SEM (n = 4). (F, G) Differential trafficking and release of Tat47-57, P3 and P6. HeLa cells transfected with the indicated RFP fusion protein were treated with 5 µmol/L Tat47-57, P3 or P6 for 1 and 2 h before being washed and imaged by confocal microscopy. Colocalization of Tat47-57, P3 and P6 with RFP-Rab5 or RFP-Rab7 is obtained by confocal microscopy (F). HeLa cells were treated with 5 µmol/L Tat47-57, P3 or P6 for 2 h. Colocalization of Tat47-57, P3 and P6 with LysoTracker is obtained by confocal microscopy (G). Scale bars are 20 µm. ***P < 0.001, 0.001 ≤ **P < 0.01, 0.01 ≤ *P < 0.05 vs. Tat.

Figure 3 Delivery of phosphopeptides and avidin into living HeLa cells. (A) Live-cell confocal microscopy images of GpTEEI, Tat-GpTEEI conjugate, P3-GpTEEI conjugate and P6-GpTEEI conjugate uptake by HeLa cells (5 µmol/L, 2 h). (B) Cellular uptake of GpTEEI, Tat-GpTEEI conjugate, P3-GpTEEI conjugate and P6-GpTEEI conjugate (5 µmol/L, 2 h) in HeLa cells. ***P < 0.001, 0.001 ≤ **P < 0.01, 0.01 ≤ *P < 0.05 vs. Tat-GpTEEI. Results are the mean ± SEM (n = 4). (C) Co-localization of FITC labeled Tat-GpTEEI conjugate, P3-GpTEEI conjugate and P6-GpTEEI conjugate (5 µmol/L, 2 h) and lysosome in HeLa cells. (D) Live-cell confocal microscopy images of avidin, TatD-avidin conjugate, P3D-avidin conjugate and P6D-avidin conjugate uptake by HeLa cells (5 µmol/L, 2 h). (E) Cellular uptake of avidin, TatD-avidin conjugate, P3D-avidin conjugate and P6D-avidin conjugate (5 µmol/L, 2 h) in HeLa cells. ***P < 0.001, 0.001 ≤ **P < 0.01, 0.01 ≤ *P < 0.05 vs. TatD & avidin. Results are the mean ± SEM (n = 4). (F) Co-localization of FITC labeled TatD-avidin conjugate, P3D-avidin conjugate and P6D-avidin (5 µmol/L, 2 h) conjugate and lysosome in HeLa cells. Images were obtained by live-cell confocal microscopy. Scale bar = 20 µm.
Table 1. Investigated peptides, their purity and molecular weight.
| Entry | Name | Sequence | Position | Purity (%) | Theoretical MW | Measured MW |
| 1 | Tat | FITC-Acp-YGRKKRRQRRR-NH2 | None | 99.17 | 2060.09 | 2061.11 |
| 2 | P1 | |
1+8 | 99.95 | 2268.95 | 2270.56 |
| 3 | P2 | |
2+9 | 98.43 | 2346.95 | 2348.56 |
| 4 | P3 | |
4+8 | 99.96 | 2303.92 | 2305.61 |
| 5 | P4 | |
4+11 | 99.94 | 2275.88 | 2277.48 |
| 6 | P5 | |
5+9 | 99.67 | 2275.88 | 2277.48 |
| 7 | P6 | |
5+12 | 99.96 | 2431.98 | 2432.95 |
| 8 | uP3 | FITC-Acp-YGRCKRRCRRR-NH2 | None | 95.43 | 2009.95 | 2010.96 |
| 9 | P3a | |
4+8 | 99.84 | 2155.93 | 2156.91 |
| 10 | P3b | |
4+8 | 99.48 | 2188.03 | 2189.04 |
| 11 | P3c | |
4+8 | 99.69 | 2007.94 | 2008.95 |
| 12 | P3d | |
None | 99.01 | 2348.98 | 2349.94 |
| 13 | P3e | |
None | 96.47 | 2349.01 | 2349.97 |
| 14 | uP6 | FITC-Acp-YGRKCRRQRRRCRR-NH2 | None | 97.90 | 2431.98 | 2432.95 |
| 15 | P6b | |
5+12 | 99.48 | 2316.09 | 2317.09 |
| 16 | P6c | |
5+12 | 99.48 | 2138.01 | 2139.02 |
| 17 | P6d | |
None | 99.45 | 2348.98 | 2349.95 |
| 18 | P6e | |
None | 96.90 | 2477.07 | 2478.03 |
| ||||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载: