Enantioselective O-H Bond Insertion of α-Diazoketones with Alcohols Cooperatively Catalyzed by Achiral Dirhodium Complexes and Chiral Spiro Phosphoric Acids
Received Date:
15 June 2018 Available Online:
15 November 2018
Fund Project:
Project supported by the National Natural Science Foundation of China (Nos. 21625204, 21532003, 21421062), the "111" Project of the Ministry of Education of China (No. B06005), the National Program for Special Support of Eminent Professionals and the Fundamental Research Funds for the Central Universities
† These authors contributed equally to this work. Supporting information for this article is available free of charge via the Internet at http://sioc-journal.cn.
Abstract:
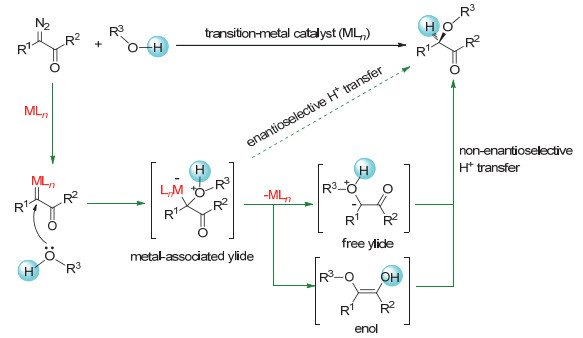
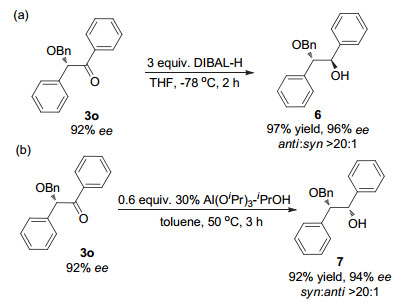
Transition-metal-catalyzed asymmetric insertion of carbene into O-H bonds is a straightforward method for the synthesis of chiral alcohols and their derivatives. In recent years, a variety of chiral catalysts have been developed to achieve high enantioselective insertions of metal carbenes derived from α-diazoesters into O-H bonds of alcohols, phenols, carboxylic acids, and even water. However, there are few successful examples of the asymmetric O-H bond insertion using α-diazoketones as carbene precursors. In this paper, we report the first asymmetric O-H insertion of α-diazoketones with alcohols co-catalyzed by achiral dirhodium complexes and chiral spiro phosphoric acids. The reaction has high yields and high enantioselectivity (up to 95% ee). The present O-H bond insertion reaction provides an efficient method for the synthesis of very useful chiral α-alkoxy ketones, which are easily transformed to corresponding 1, 2-diol derivatives with excellent diastereoselectivity. The density functional theory (DFT) calculation was performed to study the mechanism of the reaction. It is found that the chiral spiro phosphoric acid can promote the proton transfer process of enol intermediates generated from rhodium carbene and alcohol like chiral proton-transfer shuttle and realize enantioselectivity control accordingly. Water are likely to participate in this proton transfer step and has a remarkable effect on the enantiocontrol of the reaction. A typical procedure for the enantioselective O-H bond insertion of α-diazoketones is as follows. Powered Rh2(TPA)4 (2.9 mg, 0.002 mmol, 1 mol%) and chiral spiro phosphoric acid (R)-1k (3.3 mg, 0.004 mmol, 2 mol%) were introduced into an oven-dried Schlenk tube in an argon-filled glovebox. After CHCl3 (2 mL) was injected into the Schlenk tube, the solution was stirred at 25℃ under the argon atmosphere. A solution of benzyl alcohol (21.6 mg, 0.2 mmol) and 1-diazo-1-phenylpropan-2-one (2a, 33.8 mg, 0.21 mmol) in 1 mL of CHCl3 were then introduced into the Schlenk tube containing catalysts. The resulting mixture was stirred at 25℃ until the diazo compound 2a disappeared. After concentration in vacuo, the residue was purified by flash chromatography on silica gel (petroleum ether/ethyl acetate, V:V=15:1) to give (-)-1-(benzyloxy)-1-phenyl-propan-2-one (4a, 43.2 mg, 0.18 mmol, 90% yield) as a colorless oil.
a Reaction conditions: 0.002 mmol of [Rh], 0.004 mmol of SPA, 0.21 mmol of 2a, 0.2 mmol BnOH, in 3 mL of CHCl3 at 25 ℃. All the reactions accomplished in 5 min. b Yield of isolated product. c Determined by HPLC. d Use 0.1 mol% Rh2(TPA)4. e Use 0.1 mol% Rh2(TPA)4 and 0.2 mol% SPA.
a Reaction conditions: 0.002 mmol of [Rh], 0.004 mmol of SPA, 0.21 mmol of 2, 0.2 mmol of BnOH, in 3 mL of CHCl3 at 25 ℃. b Yield of isolated product. c Determined by HPLC. d 0.01 mmol of [Rh], 0.02 mmol of SPA, 1.1 mmol of 2, 1.0 mmol of BnOH, in 15 mL of CHCl3 at 25 ℃. e Used 1 mol% Rh2(TPA)4 and 2 mol% (S)-1j as co-catalysts.
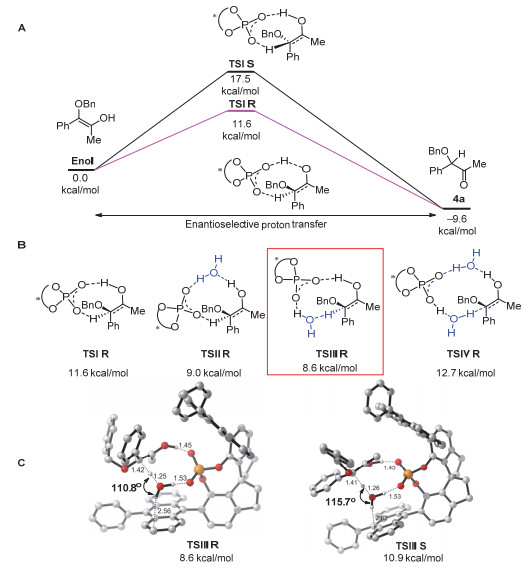
Figure 1.
(A) Calculated Gibbs energy profile for the proton-transfer of the enol, (B) water-associated proton-transfer model and (C) transition-states for R and S products (All values are Gibbs free energy)
For preparation and applications of α-diazoketones in catalytic asymmetric reactions, see: (a) Doyle, M. P.; McKervey, M. A.; Ye, T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds, Wiley, New York, 1998. (b) Doyle, M. P.; Eismont, M. Y.; Zhou, Q.-L. Russ. Chem.Bull. 1997, 46, 955. (c) Kitagaki, S.; Anada, M.; Kataoka, O.; Matsuno, K.; Umeda, C.; Watanabe, N.; Hashimoto, S. J.Am. Chem.Soc. 1999, 121, 1417. (d) Barberis, M.; Pérez-Prieto, J.; Stiriba, S.-E.; Lahuerta, P. Org.Lett. 2001, 3, 3317. (e) Hwang, C. H.; Chong, Y. H.; Song, S. Y.; Kwak, H. S.; Lee, E. Chem. Commun. 2004, 816. (f) Suga, H.; Ishimoto, D.; Higuchi, S.; Ohtsuka, M.; Arikawa, T.; Tsuchida, T.; Kakehi, A.; Baba, T. Org.Lett. 2007, 9, 4359. (g) Taber, D. F.; Tian, W. J.Org.Chem. 2008, 73, 7560. (h) Denton, J. R.; Davies, H. M. L. Org.Lett. 2009, 11, 787. (i) Xu, X.; Qian, Y.; Yang, L.; Hu, W. Chem.Commun. 2011, 47, 797. (j) Qian, Y.; Jing, C.; Liu, S.; Hu, W. Chem. Commun. 2013, 49, 2700. (k) Taber, D. F.; Paquette, C. M.; Gu, P.; Tian, W. J.Org.Chem. 2013, 78, 9772.
[5]
For non-enantioselective O—H insertion using α-diazoketones as carbene precursors, see: (a) Yates, P. J.Am.Chem.Soc. 1952, 74, 5376. (b) Shinada, T.; Kawakami, T.; Sakai, H.; Takada, I.; Ohfune, Y. Tetrahedron Lett. 1998, 39, 3757. (c) Nelson, T. D.; Song, Z. J.; Thompson, A. S.; Zhao, M.; DeMarco, A.; Reamer, R. A.; Huntington, M. F.; Grabowsk, E. J.; Reider, P. J. Tetrahedron Lett. 2000, 41, 1877. (d) Muthusamy, S.; Babu, S. A.; Gunanathan, C. Tetrahedron Lett. 2002, 43, 3133. (e) Ronan, B.; Bacqué, E.; Barrière, J. C. Tetrahedron2004, 60, 3819. (f) Muthusamy, S.; Gnanaprakasam, B.; Suresh, E. Org.Lett. 2005, 7, 4577. (g) Jung, J. C.; Avery, M. A. Tetrahedron Lett. 2006, 47, 7969.
Figure 1
(A) Calculated Gibbs energy profile for the proton-transfer of the enol, (B) water-associated proton-transfer model and (C) transition-states for R and S products (All values are Gibbs free energy)
a Reaction conditions: 0.002 mmol of [Rh], 0.004 mmol of SPA, 0.21 mmol of 2a, 0.2 mmol BnOH, in 3 mL of CHCl3 at 25 ℃. All the reactions accomplished in 5 min. b Yield of isolated product. c Determined by HPLC. d Use 0.1 mol% Rh2(TPA)4. e Use 0.1 mol% Rh2(TPA)4 and 0.2 mol% SPA.
a Reaction conditions: 0.002 mmol of [Rh], 0.004 mmol of SPA, 0.21 mmol of 2, 0.2 mmol of BnOH, in 3 mL of CHCl3 at 25 ℃. b Yield of isolated product. c Determined by HPLC. d 0.01 mmol of [Rh], 0.02 mmol of SPA, 1.1 mmol of 2, 1.0 mmol of BnOH, in 15 mL of CHCl3 at 25 ℃. e Used 1 mol% Rh2(TPA)4 and 2 mol% (S)-1j as co-catalysts.

 下载:
下载:






 下载:
下载:




 下载:
下载:
