Received Date:
27 August 2018 Available Online:
15 January 2019
Fund Project:
Project supported by the National Natural Science Foundation of China (Nos. 21320102005, 21774026) and Ministry of Science and Technology of the People's Republic of China (2016YFA0203301)
Abstract:
The dendrimers are a unique class of branched macromolecules with defined architectures synthesized by iterative reaction steps. Because of their highly branched structures, the dendrimers have a wide potential application in many fields, including sensing, drug delivery, catalysis, etc. In order to understand the thermal equilibrium behavior of the dendritic homopolymers in solution, we derived the self-consistent field theory (SCFT) for the dilute dendrimer solutions. The center segment is anchored on the origin of the space, and the shape of the dendrimer is assumed to be spherically symmetric. The pre-averaged interaction parameter u is employed to represent the volume exclusion interaction between the segments. We only focus on the dendrimer immersed in the θ solvent, where the volume exclusion interaction between the segments is negligible (u=0). The number density of the segments, φ(r), is calculated via systematically changing the topological parameters of the molecule, including the functionality f0 of the central segment, the functionality f of the branching points, the degree of polymerization of the spacers P, and the total generation number G. With all parameter combinations, φ(r) was found always maximized at the center and monotonically decreasing along the radial direction. Thus, the dendrimers in θ solvent obeys the "dense-core" model instead of the "dense-shell" model. Increasing f0, f and G results in the increase of φ(r) with any radius r. However, increasing P causes the decrease of φ(r) near the center region and the increase of φ(r) with larger r. The size of the dendrimer, analyzed by calculating the radius of gyration R, increases with f0, f, G and P. R calculated by our SCFT agrees well with the results obtained by the Rouse dynamics. With large f0, f and G, both SCFT and the Rouse dynamics predict the scaling law <R2>≈GPa2.
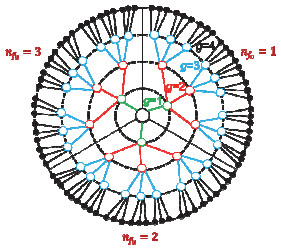
Figure 1.
Sketch of the structure of a dendrimer with G=4, P=1, f0=3, and f=4. The circles and the black spots represent the branch segments and the free end segments, respectively. The lines with different colors denotes the spacers with different generation number g
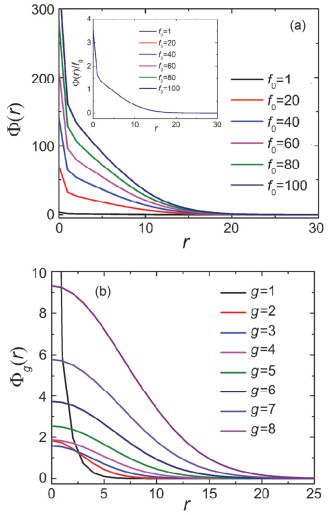
Figure 2.
The segment number density profile Φ(r) of dendrimer with different f0 and the insert shows the curves of Φ(r)/f0 in Figure (a). The figure (b) shows the segment number density profile Φg(r) with f0=20. The parameters are G=8, f=3 and P=20
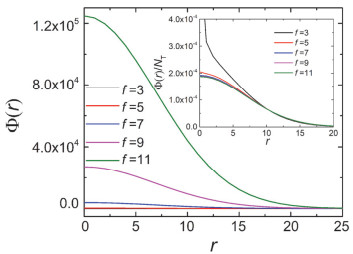
Figure 3.
The segment density profile Φ(r) of the dendrimers with different functionality f of the branch segment in the θ solvent. The insert shows the curves of Φ(r)/NT. The parameters are G=8, f0=3 and P=20
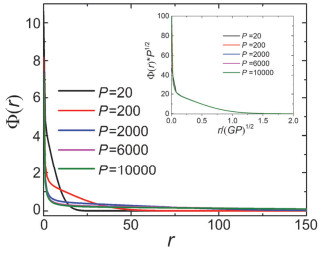
Figure 4.
The segment number density profile Φ(r) of the dendrimers with different spacer polymerization number P. The insert shows the curves Φ(r) with the x-coordinate r/(GP)1/2and y-coordinate Φ(r)·P1/2. The parameters are G=8, f0=3 and f=3
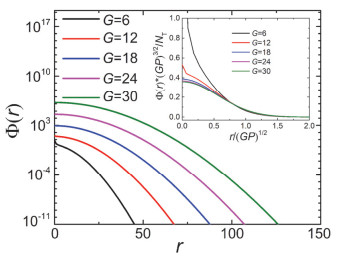
Figure 5.
The segment number density profile Φ(r) of the dendrimers with different total generation number G. The insert shows the curves of Φ(r) with the x-coordinate r/(GP)1/2 and y-coordinate Φ(r)·(GP)3/2/NT. The other parameters are f0=3, f=3 and P=20
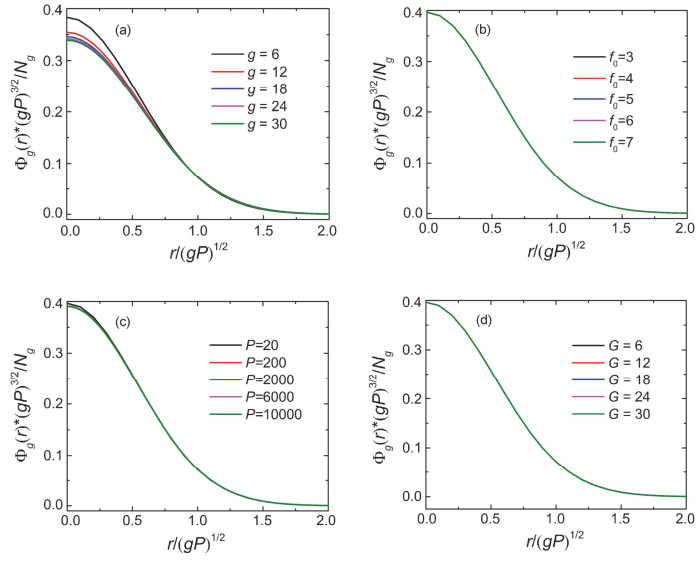
Figure 6.
(a) The segment number density profile Φg(r)·(gP)3/2/Ng of different generation spacer g=6~30 of the dendrimers with total generation number G=30. The parameters are f0=3, f=3 and P=20. (b), (c) and (d) Φg(r)·(gP)3/2/Ng of the g=5 spacer of the dendrimers with different f0, P and G, respectively. The parameters are (b) f=3, P=20, G=8; (c) f0=3, f=3, G=8; (d) f0=3, f=3, P=20
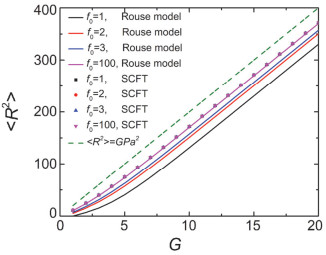
Figure 7.
The mean squared radius of gyration < R2 > , calculated by SCFT and Rouse model, as the function of G with different f0. The dash line is < R2 > =GPa2. The parameters are G=8, f=3 and P=20
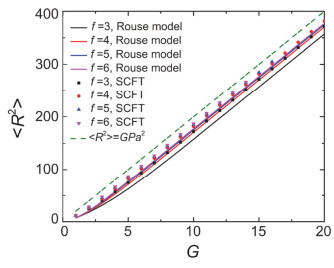
Figure 8.
The mean squared radius of gyration < R2 > , calculated by SCFT and Rouse model, as the function of G with different f. The dash line is < R2 > =GPa2. The parameters are G=8, f0=3, P=20
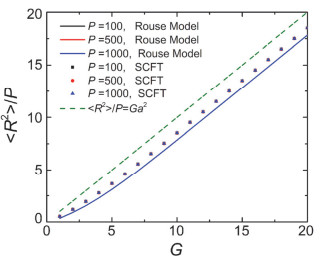
Figure 9.
The mean squared radius of gyration < R2 > /P, calculated by SCFT and Rouse model, as the function of G with different P. The dash line is < R2 > /P=Ga2. The parameters are G=8, f0=3 and f=3
Adachi, N.; Sugiyama, H.; Arai, M.; Ogawa, H. Molecules2014, 19, 4135; doi: 10.3390/molecules19044135
[5]
Shaw, P. E.; Cavaye, H.; Chen, S. S. Y.; James, M.; Gentle, I. R.; Burn, P. L. Phys. Chem. Chem. Phys.2013, 15, 9845. doi: 10.1039/c3cp51372f
[6]
Rahman, M. A.; Noh, H. B.; Shim, Y. B. Anal. Chem. 2008, 80, 8020 doi: 10.1021/ac801033s
[7]
Peng, X. H.; Pan, Q. M.; Rempel, G. L. Chem. Soc. Rev.2008, 37, 1619. doi: 10.1039/b716441f
[8]
Yoon, H. C.; Hong, M. Y.; Kim, H. S. Anal. Chem. 2000, 72, 4420. doi: 10.1021/ac0003044
[9]
Bielinska, A. U.; Yen, A.; Wu, H. L.; Zahos, K. M.; Sun, R.; Weiner, N. D.; Baker, J. R.; Roessler, B. J. Biomaterials2000, 21, 877. doi: 10.1016/S0142-9612(99)00229-X
[10]
Vincent, L.; Varet, J.; Pille, J.-Y.; Bompais, H.; Opolon, P.; Maksimenko, A.; Malvy, C.; Mirshahi, M.; Lu, H.; Vannier, J.-P.; Soria, C.; Li, H. Int. J. Cancer2003, 105, 419. doi: 10.1002/(ISSN)1097-0215
[11]
Jain, S.; Kaur, A.; Puri, R.; Utreja, P.; Jain, A.; Bhide, M.; Ratnam, R.; Singh, V.; Patil, A. S.; Jayaraman, N.; Kaushik, G.; Yadav, S.; Khanduja, K. L. Eur. J. Med. Chem.2010, 45, 4997. doi: 10.1016/j.ejmech.2010.08.006
Kumar, A.; Biswas, P. Macromolecules2010, 43, 7378. doi: 10.1021/ma101142z
[31]
Cui, W.; Su, C. F.; Merlitz, H.; Wu, C. X.; Sommer, J. U. Macromolecules2014, 47, 3645. doi: 10.1021/ma500129h
[32]
Bosko, J. T.; Prakash, J. R. Macromolecules2011, 44, 660. doi: 10.1021/ma102094f
[33]
Rubio, A. M.; McBride, C.; Freire, J. J. Macromolecules2014, 47, 5379. doi: 10.1021/ma501127f
[34]
Mandal, T.; Dasgupta, C.; Maiti, P. K. J. Chem. Phys.2014, 141, 144901. doi: 10.1063/1.4897160
[35]
Lu, Y.; An, L.; Wang, Z. G. Macromolecules2013, 46, 5731. doi: 10.1021/ma400872s
[36]
Jana, C.; Jayamurugan, G.; Ganapathy, R.; Maiti, P. K.; Jayaraman, N.; Sood, A. K. J. Chem. Phys.2006, 124, 204719. doi: 10.1063/1.2194538
[37]
Porcar, L.; Hong, K.; Butler, P. D.; Herwig, K. W.; Smith, G. S.; Liu, Y.; Chen, W. R. J. Phys. Chem. B2010, 114, 1751. doi: 10.1021/jp9064455
[38]
Prosa, T. J.; Bauer, B. J.; Amis, E. J. Macromolecules2001, 34, 4897. doi: 10.1021/ma0002186
[39]
Pötschke, D.; Ballauf, M.; Lindner, P.; Fischer, M.; Vögtle, F. J. Appl. Crystallogr.2000, 33, 605. doi: 10.1107/S0021889899013795
[40]
Rosenfeldt, S.; Dingenouts, N.; Ballauf, M.; Werner, N.; Vögtle, F.; Lindner, P. Macromolecules2002, 35, 8098. doi: 10.1021/ma020585c
[41]
Prosa, T. J.; Bauer, B. J.; Amis, E. J.; Tomalia, D. A.; Scherrenberg, R. J. Polym. Sci. Part B Polym. Phys.1997, 35, 2913. doi: 10.1002/(ISSN)1099-0488
Figure 1
Sketch of the structure of a dendrimer with G=4, P=1, f0=3, and f=4. The circles and the black spots represent the branch segments and the free end segments, respectively. The lines with different colors denotes the spacers with different generation number g
Figure 2
The segment number density profile Φ(r) of dendrimer with different f0 and the insert shows the curves of Φ(r)/f0 in Figure (a). The figure (b) shows the segment number density profile Φg(r) with f0=20. The parameters are G=8, f=3 and P=20
Figure 3
The segment density profile Φ(r) of the dendrimers with different functionality f of the branch segment in the θ solvent. The insert shows the curves of Φ(r)/NT. The parameters are G=8, f0=3 and P=20
Figure 4
The segment number density profile Φ(r) of the dendrimers with different spacer polymerization number P. The insert shows the curves Φ(r) with the x-coordinate r/(GP)1/2and y-coordinate Φ(r)·P1/2. The parameters are G=8, f0=3 and f=3
Figure 5
The segment number density profile Φ(r) of the dendrimers with different total generation number G. The insert shows the curves of Φ(r) with the x-coordinate r/(GP)1/2 and y-coordinate Φ(r)·(GP)3/2/NT. The other parameters are f0=3, f=3 and P=20
Figure 6
(a) The segment number density profile Φg(r)·(gP)3/2/Ng of different generation spacer g=6~30 of the dendrimers with total generation number G=30. The parameters are f0=3, f=3 and P=20. (b), (c) and (d) Φg(r)·(gP)3/2/Ng of the g=5 spacer of the dendrimers with different f0, P and G, respectively. The parameters are (b) f=3, P=20, G=8; (c) f0=3, f=3, G=8; (d) f0=3, f=3, P=20
Figure 7
The mean squared radius of gyration < R2 > , calculated by SCFT and Rouse model, as the function of G with different f0. The dash line is < R2 > =GPa2. The parameters are G=8, f=3 and P=20
Figure 8
The mean squared radius of gyration < R2 > , calculated by SCFT and Rouse model, as the function of G with different f. The dash line is < R2 > =GPa2. The parameters are G=8, f0=3, P=20
Figure 9
The mean squared radius of gyration < R2 > /P, calculated by SCFT and Rouse model, as the function of G with different P. The dash line is < R2 > /P=Ga2. The parameters are G=8, f0=3 and f=3

 下载:
下载:

 下载:
下载:










 下载:
下载:
