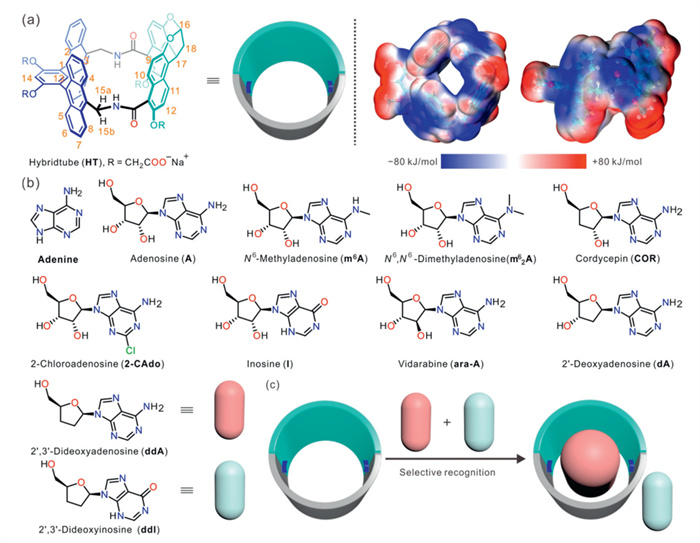
Figure 1.
(a) Chemical structure and the electrostatic potentials of HT. (b) Chemical structures of A and its analogs in this work. (c) Schematic illustration of substrate and product recognition by HT.
Hybridtube with endo-functionalized cavity for highly selective recognition: Subtle structure changes in guest lead to significant binding affinity variations
Yan-Fang Wang , Jia Liu , Wei Li , Song-Meng Wang , Jian Qin , Cheng-Da Zhao , Liu-Pan Yang , Li-Li Wang

Molecular recognition, as the cornerstone of the operation of biological systems, is extensively involved in core life processes, including cellular signal transduction, immune responses, and gene expression regulation [1]. Within organisms, the specific binding between receptors and substrates, such as the precise docking of antibodies with antigens and the efficient catalysis by enzymes with their substrates, relies critically on highly specific molecular recognition mechanisms [2,3]. Biological receptors typically achieve specific recognition of target substrates through non-covalent interactions and hydrophobic effects within their hydrophobic pockets [4]. Even when confronting structurally similar competitive substrates, they can effectively distinguish targets by leveraging unique recognition patterns.
Nevertheless, applying this natural molecular recognition mechanism to the design and construction of biomimetic receptors poses significant challenges [5,6]. The aqueous-phase recognition ability of biomimetic receptors often struggles to achieve both selectivity and affinity, especially when simultaneously recognizing guests with extremely similar structures [7]. This is because biomimetic receptors typically cannot precisely replicate the intricate three-dimensional architecture of protein binding pockets, thereby hindering the precise orchestration of multiple noncovalent interactions simultaneously.
Consequently, introducing polar binding sites into the hydrophobic cavities of biomimetic receptors enables the synergistic utilization of hydrophobic effects and noncovalent interactions, thereby achieving high binding affinity and selectivity [8–10]. Among these, noncovalent interactions such as hydrogen bonding and salt bridges can directionally bind target guest molecules, significantly enhancing both binding affinity and recognition selectivity. Building upon this principle, the strategy of directional functional group modification on the inner walls of hydrophobic cavities has been widely employed in the design and synthesis of highly selective artificial receptors. For instance, Ballester's water-soluble calix[4]pyrrole achieved the recognition of pyridine-N-oxides and their derivatives in water through N–H groups modified on its lower rim [11,12]. Davis et al. realized selective recognition of d-glucose by incorporating multiple hydrogen bonding sites within a molecular cage [13]. Jiang et al. utilized a simultaneous construction strategy to introduce N–H groups between two bisnaphthalene clefts, enabling selective recognition and detection of dioxane [14,15], and achieved efficient recognition of organic carboxylic acids via deeply embedded salt bridges within a naphthotube [16,17]. Recently, they also designed anthotube based on a bend anthracene dimer for selective recognition of quinone derivatives [18], and further realized selective recognition toward neutral guests through stepwise substitution of phenyl groups in the anthotube with pyridyl groups [19]. Additionally, Leigh/Smith et al. reported the selective recognition of guests in aqueous phase by tetralactam macrocycles [20,21]. Our group has reported naphthalene-containing tetralactam macrocycles for the selective recognition of nucleoside compounds [22], along with their applications in adsorption separation, pollutant removal, and biosensing detection [23–25].
Recently, we synthesized a novel amide hybridtube (HT) (Fig. 1a) with an electron-rich cavity by integrating a bisnaphthalene cleft with a bend anthracene dimer [26]. The precise recognition of pyranone compounds has been achieved through the strict adjustment of the distance between the inner modification sites by exploiting the structural characteristics of a rigid skeleton. More significantly, HT maintains high binding affinity while exhibiting enhanced selectivity towards structurally analogous guests. Given that adenosine (A) possesses a size comparable to pyranones and features abundant hydrogen bonding sites, we hypothesized that HT could also selectively recognize A and its analogs in water. A, an endogenous purine nucleoside, plays critical roles in processes such as vascular function, immune escape, and tumor growth. By modifying its adenine ring or ribose ring, structurally diverse A analogs can be generated [27], which demonstrate significant clinical value for treating various diseases, including inflammation, arrhythmia, and diabetes (Fig. 1b) [28]. In some cases, they exhibit superior receptor selectivity and bioactivity compared to A itself. Consequently, recognizing A and its analogs is crucial for understanding their roles in physiological and pathological processes and forms the basis for developing novel drugs. Previous studies predominantly focused on selectively distinguishing A from other ribonucleosides (G, U, and C) [29,30]. However, recognizing and differentiating A and its analogs presents a greater challenge due to their minute structural differences. Although Liu and co-workers achieved highly specific recognition of A over 2′-deoxyadenosine (dA) by incorporating boronic acid and aptamers within molecularly imprinted polymers [31], to date, no artificial receptor capable of simultaneously fulfilling the requirements of both high affinity and high selectivity for recognizing A and its analogs has been reported [32–34].
Herein, we thoroughly investigated the aqueous-phase selective recognition behavior of HT towards A and its analogs. Capitalizing on its distinct selectivity differences for 2′,3′-dideoxyadenosine (ddA), 2′,3′-dideoxyinosine (ddI), and the dye crystal violet (CV), we implemented a substrate-selective fluorescence supramolecular tandem assay (STA) for detecting the activity of adenosine deaminase (ADA) (Fig. 1c).
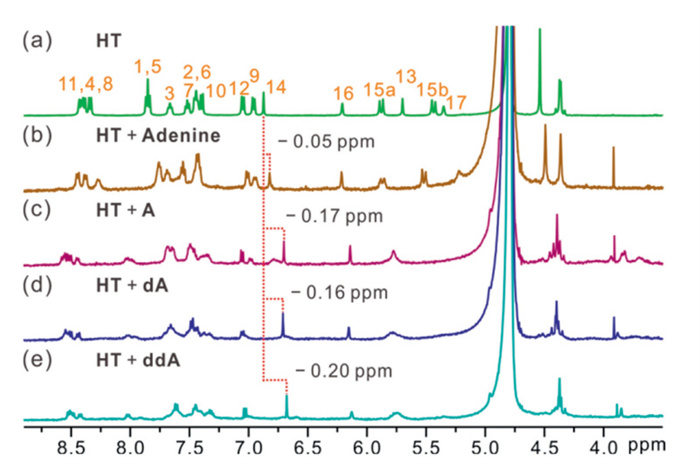
In order to study the selective recognition ability of HT, the complexation of HT to A and its analogs was tested by using 1H NMR spectroscopy in deuterated water first. As shown in Fig. 2, when HT is mixed with adenine at a 1:1 molar ratio, no significant chemical shifts were observed for the host or guest signals in the 1H NMR spectrum. In contrast, upon equimolar concentration of A, dA, or ddA mixed with HT, respectively, the aromatic proton 14 of HT in the 1H NMR spectrum was significantly upfield shifted compared to that of the free host. The proton signals of other glucosides also undergo obvious NMR chemical shifts upon the addition of an equimolar concentration of HT (Figs. S1-S11 in Supporting information). These results indicate that the HT prefers to bind glucosides rather than single bases.
The accurate binding constants of HT to the selected guests were then evaluated by fluorescence (FL) titration (Figs. S12-S22 in Supporting information), and the data were listed in Table 1. Adenine possessed a decent binding constant with HT at a value of 4.07 × 103 L/mol. However, A with an additional ribose group possessed a much stronger binding constant (5.98 × 104 L/mol). We speculated that the introduced ribose ring could lead to the increased volume to fill in the hydrophobic cavity, which facilitates the discharge of more "cavity water". Consequently, the hydrophobic effect can be enhanced and result in a stronger binding constant. With one or two methyl group modifications on the nucleobases to obtain N6-methyladenosine (m6A) or N6, N6-dimethyladenosine (m26A), the binding constant further increased to 1.57 × 105 or 3.01 × 106 L/mol, respectively. The methylation of nucleoside increased its hydrophobicity and thus enhances the binding strength. The binding constant of 2-chloroadenosine (2-CAdo) was as same as A. Though replacing the H by Cl at the 2-position made the 2-CAdo possess lower water-solubility than A, the decreased size matched-degree counteracted the increased hydrophobic effect. When the amino group of A is changed to a carbonyl group, HT showed a slight decrease in binding affinity to inosine (I), which may stem from the alteration of hydrogen-bonding sites.
 DownLoad:
CSV
DownLoad:
CSV
| Guest | Chemical structures | Ka (FL) |
| Adenine |  |
(4.07 ± 0.23) × 103 |
| A |  |
(5.98 ± 0.18) × 104 |
| m6A |  |
(1.57 ± 0.11) × 105 |
| m26A |  |
(3.01 ± 0.26) × 106 |
| 2-CAdo |  |
(5.99 ± 0.19) × 104 |
| I |  |
(2.54 ± 0.28) × 104 |
| ara-A |  |
(1.10 ± 0.22) × 105 |
| dA |  |
(3.69 ± 0.46) × 106 |
| COR |  |
(4.50 ± 0.22) × 105 |
| ddA |  |
(2.21 ± 0.35) × 106 |
| ddI |  |
(1.71 ± 0.11) × 105 |
In addition to the changes on the purine ring, minor modifications on the ribose moiety can also significantly affect the binding constants. As shown in Table 2, the dehydroxylation of ribose at the 2′-position led to a remarkable enhancement in the binding (A/dA up to 61.7-fold). Additionally, the migration of the hydroxyl group from the 2′-position to the 3′-position of ribose also led to an enhanced binding affinity (COR/dA up to 8.2-fold). The binding affinities of HT were significantly affected by minor structural differences, whether in base or ribose. However, the previously reported macrocycles were only capable of recognizing changes in the number of hydroxy groups on the ribose ring, but not the alterations in the positions of these hydroxy groups (Figs. S26-S28 in Supporting information and Table 2). The findings demonstrate that artificial macrocycles featuring rigid internally modified cavities exhibit significant advantages in achieving highly selective recognition. In contrast, macrocycles with flexible internally modified cavities can adjust their conformations to alter hydrogen-bonding distances, matching various guest molecules, which consequently results in relatively lower selectivity. Furthermore, classical macrocycles lacking internal modification sites exhibit significantly compromised binding affinity and selectivity due to their inability to establish directional interactions.
DownLoad:
CSV
| Guests | Hosts | |||
| HT | TM | CB7 | β-CD | |
| A | (5.98 ± 0.18) × 104 a | (6.48 ± 0.30) × 103 b [32] | 8.06 × 103 b [33] | 13.3 c [34] |
| dA | (3.69 ± 0.46) × 106 a | (1.82 ± 0.30) × 104 b | 5.62 × 104 b [33] | 25.1 c [34] |
| COR | (4.50 ± 0.22) × 105 a | (1.43 ± 0.08) × 104 b | 1.64 × 104 b [33] | 25.2 d [34] |
| KA/KdA/KCOR | 1:61.7:7.5 | 1:2.8:2.2 | 1:7.0:2.0 | 1:1.9:1.9 |
| a Measured by FL titration. b Measured by ITC titration. c Measured by solubility method. d Measured by circular dichroism. |
||||
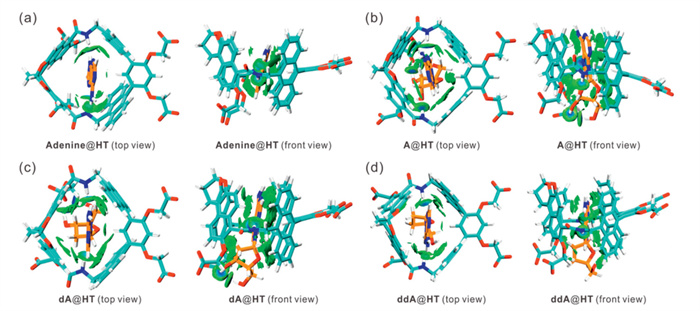
To further demonstrate the ability of HT to recognize A and its analogs effectively, we conducted density functional theory (DFT) calculations [35,36], as detailed in Fig. 3, and Figs. S29 and S30 (Supporting information). Our findings revealed that the nitrogen atoms at positions 3 and 7 of the purine ring in adenine, dA, and ddA each formed hydrogen bonds with the two amide protons of HT, indicating that N–H···N interactions are involved in the formation of the complexes (Figs. 3a, c and d, and Figs. S29a, c and d). However, in the case of A, only the nitrogen atom at position 7 of the purine ring establishes a hydrogen bond with an amide proton from HT (Fig. 3b and Fig. S29b in Supporting information). This observation suggests that the hydroxyl group at the 2′-position of the ribose ring creates a steric hindrance, which is unfavorable for forming a hydrogen bond between the nitrogen atom at position 3 of the purine ring and the amide proton of the host. Consequently, the binding affinity is reduced compared to that of dA and ddA. Additionally, the nitrogen atom at position 3 of the purine ring of ddI changed from a hydrogen bond acceptor to a hydrogen bond donor. It prevents the formation of a hydrogen bond, leading to a reduced binding strength (Fig. S30). The above results illustrate the importance of hydrogen bond binding sites in the cavity for selective guest recognition.
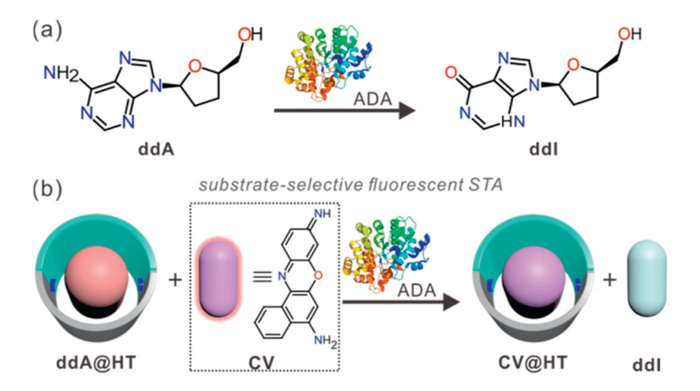
Given that HT is up to 13-fold selective for ddA and ddI (Table 1), the substrate-selective STA strategy can be used for ADA activity detection. To further evaluate the applicability of this strategy under physiologically relevant conditions, we measured the binding constants of ddA and ddI to HT in phosphate buffer (PB) at 37 ℃ (Figs. S23 and S24 in Supporting information). The calculated guest selectivity was approximately 12-fold. Compared with the results obtained in water, this selectivity did not change significantly, which corroborates the suitability of the STA-based strategy for monitoring ADA activity. ADA is a crucial enzyme involved in the metabolism of purine nucleosides both within and outside the cell, catalyzing the irreversible hydrolytic deamination of A to produce inosine (I), which plays an important role in the maturation, maintenance, and function of the immune system [37]. Abnormal levels of ADA are closely associated with the development of various diseases [38]. For instance, it is well known that ADA gene deficiency can result in severe combined immunodeficiency disease (SCID) [39–42] and neutropenia [43]. On the other hand, elevated ADA activity is associated with conditions such as tuberculous meningitis [44], atherosclerotic vessels [45], and autoimmune diseases [46]. Therefore, the sensitive and rapid detection of ADA activity is essential for the early diagnosis and therapeutic monitoring of these diseases.
Conventional methods for determining ADA enzyme activity include techniques such as ultraviolet (UV) spectroscopy [47], Berthelot colorimetry [48], high-performance liquid chromatography (HPLC) [49], capillary electrophoresis (CE) [50], and aptamer-sensor assays [51]. These methods can selectively or simultaneously detect substrates and products or measure free ammonia generated by ADA-catalyzed reactions. However, they are often plagued by low sensitivity, non-real-time detection, complex equipment requirements, and cumbersome procedures or background interference. To address these limitations, we developed a simple, sensitive, real-time method for enzyme activity monitoring and inhibitor screening, namely the STA, based on the ADA enzyme function of HT. The first step in establishing this method involves selecting a suitable fluorescent dye as the signaling element. Our previous research has demonstrated that the fluorescence signals of free phenoxazine dyes undergo significant quenching when bound to macrocycles, making them effective signaling indicators for STA sensors [25]. In this study, we constructed a substrate-selective STA by choosing CV dye as a fluorescent indicator and applying it to the ADA enzyme activity assay (Fig. 4). Initially, the substrate ddA acts as a strong competitor, replacing the fluorescent indicator in the macrocyclic cavity, which results in a strong fluorescent signal emission from the free dye. During the enzymatic reaction, the ADA enzyme catalyzes the hydrolysis and deamination of the substrate ddA, generating the ddI product. This product is a weaker competitor, allowing the CV dye to re-enter the macrocyclic cavity, quenching the dye's fluorescence. Based on the above mechanism, the activity of the ADA enzyme can be quantitatively analyzed by monitoring the fluorescence switch-off response of the fluorescent indicator CV.
Through fluorescence titration experiments combined with nonlinear fitting analysis, the association constant of CV with HT was determined to be 3.78 × 106 L/mol (Fig. S25 in Supporting information). This value is comparable to the binding constant of ddA with HT. To construct a sensitive STA, the concentration ratio of CV to HT was optimized in this study and ultimately set to 0.5 μmol/L and 2.4 μmol/L, respectively. The experimental results demonstrated that the difference in the optical response of the CV@HT complex to the substrate ddA and the product ddI was most significant at this concentration ratio (Fig. S31 in Supporting information). The fluorescence intensity of CV significantly increased after the addition of ddA to the CV@HT complex in PB buffer at 37 ℃, whereas no significant change in the fluorescence intensity of CV was observed after the addition of ddI (Figs. S31d and S32 in Supporting information), which is in agreement with the results of the selectivity determined by the titration experiments. Additionally, control experiments were performed by adding equal amounts of ddA and ddI to HT, respectively. HT itself did not exhibit any fluorescence signal at 632 nm, and adding either guest did not result in any fluorescence changes (Fig. S33 in Supporting information). The above experiments indicate that the changes in the fluorescence signal of the CV@HT reporter pair are indeed caused by the dye entering and exiting the macrocyclic cavity.
To ensure the accuracy and reliability of our experimental results, we investigated whether HT affects ADA activity using the Berthelot colorimetric method [48]. During the enzymatic reaction, the substrate ddA undergoes hydrolytic deamination, producing free ammonia. Therefore, the activity of ADA can be assessed by measuring the amount of NH3 released in the reaction system. Our experiments showed that the presence or absence of HT did not affect ammonia production from hydrolytic deamination, indicating that HT does not interfere with ADA activity (Fig. S34 in Supporting information).
In terms of experimental design, we first created several experimental groups with varying concentrations of ADA while keeping the substrate concentration of ddA constant. We monitored the change in fluorescence value over time during the enzymatic reaction for each group in real-time (Fig. S35 in Supporting information). After considering the equilibrium time of the enzymatic reaction and the detection sensitivity, we ultimately set the ADA concentration at 1 μg/mL. To further explore the enzymatic kinetic properties of ADA, we conducted experiments using a series of different ddA concentrations and observed the progress of the reactions, showing the expected dependence of the enzyme kinetics on the substrate concentration (Fig. 5a). Based on the experimental data, fitting analysis was performed according to the Michaelis-Menten kinetic model, and the Michaelis constant (KM) of ADA was obtained to be 2.12 ± 0.01 μmol/L (Fig. 5b). It is important to note that KM can be influenced by various factors such as reaction conditions, analytical methods, and enzyme sources. As a result, the KM values reported in the literature have varied significantly, ranging from 25 μmol/L to 550 μmol/L [52–54]. All of the above studies have taken a pre-incubation approach to detect enzyme activity. Comparatively, our method can realize real-time monitoring of enzyme activity while ensuring rapid detection and a low sample size. To further evaluate the practical application potential of the CV@HT sensor in enzyme activity detection, ten common biological matrix components were selected as potential interferents, including Na+, K+, Mg2+, H2PO4-, HPO42-, glycine, l-phenylalanine, ATP, creatinine, and urea. Enzymatic reactions were monitored both in PB buffer and in PB buffer containing the aforementioned interferents (Fig. S36 in Supporting information). Comparative experiments demonstrated that these interfering substances did not significantly affect the enzyme activity detection performance of the CV@HT sensor.
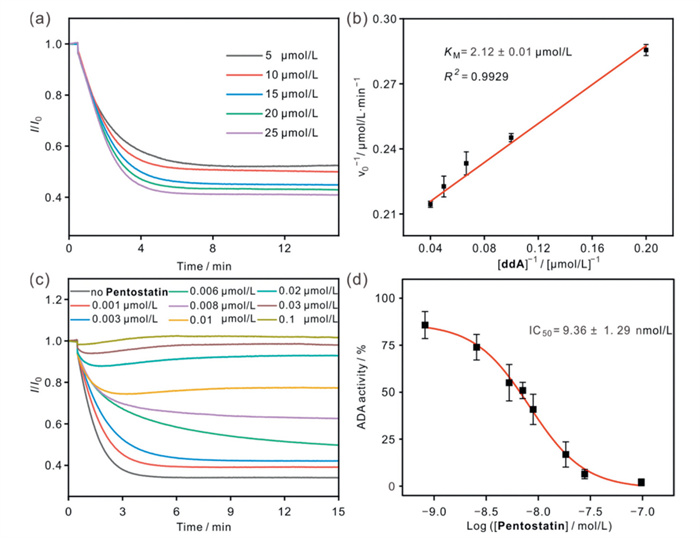
STA has been demonstrated as a powerful and straightforward method for screening enzyme inhibitors based on supramolecular reporter pairs. To assess the feasibility of STA in evaluating a variety of ADA candidate inhibitors, we investigated two representative inhibitors: the potent inhibitor Pentostatin and the weaker inhibitor Hibifolin. Our experiments revealed that neither Pentostatin nor Hibifolin significantly affected the fluorescence signals of the CV@HT reporter pair (Figs. S37 and S38 in Supporting information). This indicates that these inhibitors do not compete with the dye CV for binding to HT. Additionally, ADA activity showed a clear downward trend with increasing concentrations of Pentostatin and Hibifolin, a change that could be obtained by a sharp decrease in the initial reaction rate (Figs. 5c and d, Fig. S39 in Supporting information). The IC50 values for Pentostatin and Hibifolin on ADA were calculated to be 9.36 ± 1.29 nmol/L and 147.40 ± 10.61 μmol/L, respectively. The above results were consistent with the expected order of inhibitory strength, further confirming the effectiveness of STA in screening for ADA inhibitors.
In summary, we have demonstrated that HT, featuring endo-functionalized hydrogen bond donors, achieves highly specific recognition of A and its analogs in aqueous phase. Remarkably, it enables precise discrimination even when confronted with subtle variations in the hydrogen bond acceptors of the guests, achieving a binding selectivity as high as 61.7-fold (A/dA). This finding underscores the critical role of hydrogen bonding sites in selective molecular recognition, providing significant insights for the highly specific recognition of novel biomimetic receptors. Furthermore, we successfully constructed a fluorescence turn-off supramolecular tandem assay by utilizing HT and CV as probe components. This assay serves as a valuable tool for monitoring ADA activity and screening enzyme inhibitors, holding considerable potential for applications in disease diagnosis and drug screening.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Yan-Fang Wang: Investigation, Data curation, Conceptualization. Jia Liu: Writing – original draft, Investigation, Data curation. Wei Li: Investigation, Data curation. Song-Meng Wang: Data curation. Jian Qin: Methodology. Cheng-Da Zhao: Resources, Methodology, Investigation. Liu-Pan Yang: Writing – review & editing, Investigation, Funding acquisition. Li-Li Wang: Writing – review & editing, Supervision, Funding acquisition, Conceptualization.
This work was supported by the National Natural Science Foundation of China (Nos. 22174059 and 22571143), the Science and Technology Innovation Program of Hunan Province (No. 2022RC1230), and the Postgraduate Scientific Research Innovation Project of Hunan Province (No. CX20240853).
Supplementary material associated with this article can be found, in the online version, at doi:
E. Persch, O. Dumele, F. Diederich, Angew. Chem. Int. Ed. 54 (2015) 3290–3327. doi: 10.1002/anie.201408487
J.L. Hu, X.X. Xu, L.Q. Liu, et al., Adv. Healthcare. Mater. 14 (2025) 2404569. doi: 10.1002/adhm.202404569
W.W. Kung, S. Ramachandran, N. Makukhin, et al., Nat. Commun. 10 (2019) 2534. doi: 10.1038/s41467-019-10190-4
Q.S. Ji, Y.R. Liu, H.Y. Zhang, et al., Adv. Sci. 12 (2025) 2413185. doi: 10.1002/advs.202413185
P.S. Cremer, A.H. Flood, B.C. Gibb, D.L. Mobley, Nat. Chem. 10 (2017) 8–16.
R.W. Tang, Y.P. Ye, S.J. Zhu, et al., Chin. Chem. Lett. 34 (2023) 107734. doi: 10.1016/j.cclet.2022.08.014
F.Y. Chen, R. Fu, Z.H. Gong, et al., Angew. Chem. Int. Ed. 64 (2025) e202500916. doi: 10.1002/anie.202500916
L. Adriaenssens, P. Ballester, Chem. Soc. Rev. 42 (2013) 3261–3277. doi: 10.1039/c2cs35461f
C.D. Zhao, H. Yao, S.Y. Li, et al., Chin. Chem. Lett. 35 (2024) 108879. doi: 10.1016/j.cclet.2023.108879
J. Liu, L.H. Yi, Y.X. He, et al., Microchem. J. 217 (2025) 114878. doi: 10.1016/j.microc.2025.114878
D. Hernández-Alonso, S. Zankowski, L. Adriaenssens, P. Ballester, Org. Biomol. Chem. 13 (2015) 1022–1029. doi: 10.1039/C4OB02108H
B. Verdejo, G. Gil-Ramirez, P. Ballester, J. Am. Chem. Soc. 131 (2009) 3178–3179. doi: 10.1021/ja900151u
R.A. Tromans, S.K. Samanta, A.M. Chapman, A.P. Davis, Chem. Sci. 11 (2020) 3223–3227. doi: 10.1039/c9sc05406e
G.B. Huang, S.H. Wang, H. Ke, et al., J. Am. Chem. Soc. 138 (2016) 14550–14553. doi: 10.1021/jacs.6b09472
H. Yao, H. Ke, X.B. Zhang, et al., J. Am. Chem. Soc. 140 (2018) 13466–13477. doi: 10.1021/jacs.8b09157
X. Huang, X.P. Wang, M. Quan, et al., Angew. Chem. Int. Ed. 60 (2021) 1929–1935. doi: 10.1002/anie.202012467
X.P. Wang, M. Quan, H. Yao, et al., Nat. Commun. 13 (2022) 2291. doi: 10.24963/ijcai.2022/318
H. Zhou, X.Y. Pang, X.P. Wang, et al., Angew. Chem. Int. Ed. 60 (2021) 25981–25987. doi: 10.1002/anie.202112267
S.M. Wang, H. Nian, Y.F. Wang, et al., Sci. China Chem. 68 (2024) 369–376.
A.G. Johnston, D.A. Leigh, A. Murphy, et al., J. Am. Chem. Soc. 118 (1996) 10662–10663. doi: 10.1021/ja962046r
W. Liu, A.G. Oliver, B.D. Smith, J. Am. Chem. Soc. 140 (2018) 6810–6813. doi: 10.1021/jacs.8b04155
H. Yao, J. Qin, Y.F. Wang, et al., Chin. Chem. Lett. 35 (2024) 109154. doi: 10.1016/j.cclet.2023.109154
S.Y. Li, H. Yao, H. Hu, et al., Chem. Commun. 59 (2023) 7204–7207. doi: 10.1039/d3cc01164j
Y. Qiu, S. Yu, S.M. Wang, et al., Chem. Eng. J. 495 (2024) 153143. doi: 10.1016/j.cej.2024.153143
H. Yao, S.Y. Li, H. Zhang, et al., Chem. Commun. 59 (2023) 5411–5414. doi: 10.1039/d2cc06622j
Y.F. Wang, S.M. Wang, X.B. Zhang, et al., Angew. Chem. Int. Ed. 62 (2023) e202310115. doi: 10.1002/anie.202310115
G.G. Yegutkin, D. Boison, Pharmacol. Rev. 74 (2022) 797–822.
S. Man, Y.Y. Lu, L.J. Yin, et al., Drug Discov. Today 26 (2021) 1490–1500. doi: 10.1016/j.drudis.2021.02.020
X. Gong, B. Tang, J.J. Liu, et al., Biosens. Bioelectron. 87 (2017) 858–864. doi: 10.1016/j.bios.2016.09.027
P. Wang, J.X. Liu, Y. Ma, et al., Chem. Eng. J. 394 (2020) 124931. doi: 10.1016/j.cej.2020.124931
Y.Q. Li, Z.J. Zhang, B.W. Liu, J.W. Liu, ACS Appl. Bio. Mater. 3 (2020) 2568–2576. doi: 10.1021/acsabm.9b00936
H. Yao, Z.P. Xiao, J. Guo, et al., Sci. China Chem. 68 (2025) 4264–4275. doi: 10.1007/s11426-025-2771-7
A.A. Dewantari, N. Yongwattana, P. Payongsri, et al., Molecules 25 (2020) 2045. doi: 10.3390/molecules25092045
T.X. Xiang, B.D. Anderson, Int. J. Pharm. 59 (1990) 45–55. doi: 10.1016/0378-5173(90)90063-A
J.C. Kromann, C. Steinmann, J.H. Jensen, J. Chem. Phys. 149 (2018) 104102. doi: 10.1063/1.5047273
T. Lu, F.W. Chen, J. Comput. Chem. 33 (2012) 580–592. doi: 10.1002/jcc.22885
N.H. Elowe, R. Nutiu, A. Allali-Hassani, et al., Angew. Chem. Int. Ed. 45 (2006) 5648–5652. doi: 10.1002/anie.200601695
X.J. Xing, X.G. Liu, Y. He, et al., Biosens. Bioelectron. 37 (2012) 61–67. doi: 10.1016/j.bios.2012.04.037
A. Aiuti, M.G. Roncarolo, L. Naldini, EMBO. Mol. Med. 9 (2017) 737–740. doi: 10.15252/emmm.201707573
E. Grunebaum, C. Booth, G.D.E. Cuvelier, et al., J. Allergy. Clin. Immunol. Pract. 11 (2023) 1665–1675.
M. Migliavacca, F. Barzaghi, C. Fossati, et al., Nat. Med. 30 (2024) 488–497. doi: 10.1038/s41591-023-02789-4
B. Reinhardt, O. Habib, K.L. Shaw, et al., Blood 138 (2021) 1304–1316. doi: 10.1182/blood.2020010260
V.H.D. Kim, A. Pham-Huy, E. Grunebaum, J. Allergy. Clin. Immunol. 143 (2019) 403–405. doi: 10.1016/j.jaci.2018.04.029
A. Pormohammad, S.M. Riahi, M.J. Nasiri, et al., J. Infect. 74 (2017) 545–554.
B. Kutryb-Zajac, L. Mateuszuk, P. Zukowska, et al., Cardiovasc. Res. 112 (2016) 590–605. doi: 10.1093/cvr/cvw203
Z.W. Gao, X. Wang, H.Z. Zhang, et al., Autoimmun. Rev. 20 (2021) 102709.
J. Lu, D.G. Grenache, Clin. Chim. Acta 413 (2012) 1637–1640.
L. Silva Dalsasso Joaquim, N. Granzotto, L.F. Dos Santos, et al., J. Clin. Lab. Anal. 33 (2019) e22823.
H. Ni, Y.H. Li, R.L. Hao, et al., Int. J. Food Sci. Tech. 51 (2016) 1168–1176. doi: 10.1111/ijfs.13064
Y.F. Qi, Y.X. Li, J.J. Bao, Anal. Biochem. 506 (2016) 31–44.
J.H. Zhu, L.P. Mei, A.J. Wang, et al., Biosens. Bioelectron. 226 (2023) 115141.
P.T. Lin, S.L. Zhao, X. Lu, et al., J. Sep. Sci. 36 (2013) 2538–2543. doi: 10.1002/jssc.201300315
L. Pei, L.J. Xie, Q. Lin, et al., Anal. Biochem. 414 (2011) 131–137.
S.L. Qi, H.D. Guan, G. Deng, et al., J. Pharm. Biomed. Anal. 153 (2018) 175–181.

Figure 1 (a) Chemical structure and the electrostatic potentials of HT. (b) Chemical structures of A and its analogs in this work. (c) Schematic illustration of substrate and product recognition by HT.

Figure 2 Partial 1H NMR spectra (500 MHz, D2O, 0.5 mmol/L, 298 K) of (a) HT, (b) a 1:1 mixture of adenine and HT, (c) a 1:1 mixture of A and HT, (d) a 1:1 mixture of dA and HT, and (e) a 1:1 mixture of ddA and HT.

Figure 3 Top view and front view of non-covalent interaction analysis of (a) adenine@HT, (b) A@HT, (c) dA@HT, and (d) ddA@HT. Optimized geometry of the four host-guest complexes was obtained by DFT calculations (ωB97XD/6–31G(d) in water). Non-covalent interaction analysis (IGMH, independent gradient model based on Hirshfeld partition) was realized by Multiwfn 3.8(dev) code based on the ωB97XD/6–311G+(d,p) wavefunction at optimized structures [35,36].

Figure 4 (a) The ADA-catalyzed enzymatic reaction. (b) Schematic illustration of ADA activity monitoring with a switch-off fluorescence response based on the CV@HT reporter pair.

Figure 5 (a) Determination of the KM value by monitoring ADA (1 μg/mL) activity with various concentrations of ddA (5–25 μmol/L) in the presence of CV@HT reporter pair (0.5/2.4 μmol/L, λex = 585 nm, λem = 632 nm). (b) The Lineweaver-Burk plot for ADA. (c) Continuous fluorescent assay for ADA activity inhibition by Pentostatin (0.001–0.1 μmol/L) with the CV@HT reporter pair (0.5/2.4 μmol/L, λex = 585 nm, λem = 632 nm), ADA (1 μg/mL), and ddA (20 μmol/L). (d) Dose-response curve and associated plot analysis for ADA activity inhibition by Pentostatin. The experiments were performed in PB buffer (10 mmol/L, pH 7.4) at 37 ℃.
Table 1. Association constants (Ka, L/mol) of HT with A and its analogs in water at 25 ℃ as determined by FL titration.
| Guest | Chemical structures | Ka (FL) |
| Adenine | |
(4.07 ± 0.23) × 103 |
| A | |
(5.98 ± 0.18) × 104 |
| m6A | |
(1.57 ± 0.11) × 105 |
| m26A | |
(3.01 ± 0.26) × 106 |
| 2-CAdo | |
(5.99 ± 0.19) × 104 |
| I | |
(2.54 ± 0.28) × 104 |
| ara-A | |
(1.10 ± 0.22) × 105 |
| dA | |
(3.69 ± 0.46) × 106 |
| COR | |
(4.50 ± 0.22) × 105 |
| ddA | |
(2.21 ± 0.35) × 106 |
| ddI | |
(1.71 ± 0.11) × 105 |
 下载: 导出CSV
下载: 导出CSV
Table 2. Association constants (Ka, L/mol) of HT/tetralactam macrocycle (TM)/cucurbit[7]uril (CB7)/β-cyclodextrin (β-CD) with adenosine and its analogs in water at 25 ℃ as determined by FL titration, isothermal titration calorimetry (ITC) titration, solubility method, and circular dichroism.
| Guests | Hosts | |||
| HT | TM | CB7 | β-CD | |
| A | (5.98 ± 0.18) × 104 a | (6.48 ± 0.30) × 103 b [32] | 8.06 × 103 b [33] | 13.3 c [34] |
| dA | (3.69 ± 0.46) × 106 a | (1.82 ± 0.30) × 104 b | 5.62 × 104 b [33] | 25.1 c [34] |
| COR | (4.50 ± 0.22) × 105 a | (1.43 ± 0.08) × 104 b | 1.64 × 104 b [33] | 25.2 d [34] |
| KA/KdA/KCOR | 1:61.7:7.5 | 1:2.8:2.2 | 1:7.0:2.0 | 1:1.9:1.9 |
| a Measured by FL titration. b Measured by ITC titration. c Measured by solubility method. d Measured by circular dichroism. |
||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载: