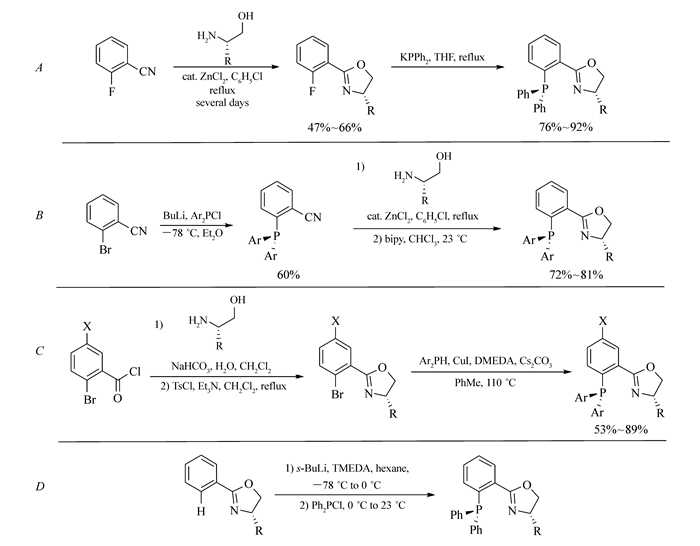
 Scheme1.
Synthesis of phosphinooxazolines(PHOX ligands) in literatures
Scheme1.
Synthesis of phosphinooxazolines(PHOX ligands) in literatures
引用本文:
曹宝辰, 吴国杰, 何宇鹏, 韩福社. 手性芳基膦-噁唑啉的简便合成[J]. 应用化学,
2018, 35(2): 189-196.
doi:
10.11944/j.issn.1000-0518.2018.02.170066

Citation: CAO Baochen, WU Guojie, HE Yupeng, HAN Fushe. A Facile Synthesis of Chiral Phosphinoaryloxazolines[J]. Chinese Journal of Applied Chemistry, 2018, 35(2): 189-196. doi: 10.11944/j.issn.1000-0518.2018.02.170066
Citation: CAO Baochen, WU Guojie, HE Yupeng, HAN Fushe. A Facile Synthesis of Chiral Phosphinoaryloxazolines[J]. Chinese Journal of Applied Chemistry, 2018, 35(2): 189-196. doi: 10.11944/j.issn.1000-0518.2018.02.170066
手性芳基膦-噁唑啉的简便合成
摘要:
膦-噁唑啉化合物作为一类"优势配体",自发现以来就引起化学家们的广泛关注。现有合成方法存在路线长、收率低、分离困难等问题。本研究发展了一种合成手性芳基膦-噁唑啉(PHOX)的简单高效方法。在1-羟基苯并三唑(HOBt)、1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDCI)作用下,2-(二苯基膦基)苯甲酸与多种光学纯的氨基醇发生缩合反应,高收率获得酰胺基醇类化合物;之后酰胺基醇经三苯基膦/三乙胺/四氯化碳体系处理完成噁唑啉环的构建,以64%~86%的总收率得到膦-噁唑啉化合物。随后,合成的化合物(S)-t-BuPHOX被应用于钯催化β-酮酯的脱羧Tsuji烯丙基化反应,得到了80%的收率和84%的ee值。该新方法具有原料易得、条件温和、收率高等优点。
-
关键词:
- 膦-噁唑啉配体
- / (二苯基膦基)苯甲酸
- / 手性氨基醇
English
A Facile Synthesis of Chiral Phosphinoaryloxazolines
Abstract:
As a widely used class of privileged ligands, phosphinoaryloxazolines(PHOX) have attracted much attention from chemists. However, the previous synthetic methods have problems of long steps, low yield and difficult separation and so on. In this article, a simple and efficient procedure for the synthesis of phosphinoaryloxazolines(PHOX) has been developed. First, 2-(diphenylphosphino)benzoic acid was condensed with various enantiomerically pure amino alcohols in the presence of 1-hydroxylbenzotriazole(HOBt) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride(EDCI) in dimethylformamide to give the corresponding (amido alcohol)s amides in excellent yields. Then the (amido alcohol)s amides were subjected to the oxazoline ring formation by treatment with triphenylphosphine, triethylamine and carbon tetrachloride in acetonitrile to afford a series of phosphinooxazolines in 64%~86% total yields. Subsequently, (S)-t-BuPHOX was applied in the palladium-catalyzed decarboxylative Tsuji allylations of β-ketoester, giving an excellent isolated yield of 80% with an enantiomeric excess of 84%. The new synthetic procedure has the advantages of using readily available starting materials, mild reaction conditions, and high overall yields.
-
1993年,Helmchen、Pfaltz和Williams几乎同时报道了手性芳基膦-噁唑啉(PHOX)作为配体在钯催化的不对称烯丙基取代反应中的应用[1-3]。之后,该类配体因结构易修饰、立体选择性高等特点被广泛应用于脱羧Tsuji烯丙化反应、Heck反应、氢化反应、[3+2]环加成反应、Diels-Alder反应等不对称催化反应,从而成为极具吸引力的“优势”配体[4-7]。由商业易得的原料出发,通过改变噁唑啉环和膦的类型可以得到一系列不同的PHOX配体[8-12]。市售光学纯的氨基醇或氨基酸可以作为手性源,亲核或亲电的磷源被用于C—P键的构建。具体来说,芳基膦-噁唑啉化合物的常用合成方法主要为以下几种:1)在ZnCl2的催化作用下,邻氟苯腈与手性氨基醇可以形成噁唑啉环,然后二苯基磷酸钾与之发生芳香亲核取代反应生成膦-噁唑啉化合物(Scheme 1A);2)邻溴苯腈经卤-锂交换之后引入膦基团,随后同样可与手性氨基醇在ZnCl2的催化作用下完成噁唑啉环的构建(Scheme 1B);3)邻溴苯甲酰氯与氨基醇发生缩合反应生成酰胺,将羟基转化为易于离去的基团之后发生环化形成噁唑啉,随后在CuI的催化作用下发生C—P键偶联反应引入膦基团(Scheme 1C);4)噁唑啉化合物在仲丁基锂/四甲基乙二胺体系作用下可以发生邻位金属化,之后与二苯基氯化膦发生取代反应从而生成目标物(Scheme 1D)。
Scheme1.
Synthesis of phosphinooxazolines(PHOX ligands) in literatures
以上方法尽管可以制备一系列芳基膦-噁唑啉化合物,但存在着一些缺点,例如关键步骤产率低、反应条件苛刻、时间长(如苯腈与氨基醇在ZnCl2作用下形成噁唑啉的反应通常需要在氯苯中高温回流数天)、操作不便(一些反应需要严格无水无氧、低温的反应条件)等。因此,发展原料易得、操作简单、高效的合成新路径就显得尤为重要。本研究以2-(二苯基膦基)苯甲酸为起始原料,与手性氨基醇反应生成羟基酰胺,随后合环生成噁唑啉环从而得到一系列膦-噁唑啉(PHOX)化合物(Scheme 2)。与已报道的方法相比,我们所发展的新方法具有原料易得,操作简单、产率高等优点。
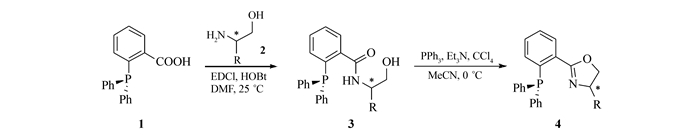 Scheme2.
New route for synthesis of phosphinooxazolines(PHOX ligands)
Scheme2.
New route for synthesis of phosphinooxazolines(PHOX ligands)
1 实验部分
1.1 仪器和试剂
AV 300、400型核磁共振仪(德国Bruker公司),TMS为内标,CDCl3为溶剂;Ultima7.0T型傅里叶变换离子回旋共振质谱仪(美国IonSpec公司),LC-20AD型高效液相色谱仪(日本SHIMAZDZU公司),色谱柱为ChiralPak AS-H(日本DAICEL Chemical公司)。
所用的2-(二苯基膦基)苯甲酸(纯度98%)、D-苯甘氨醇(纯度99%)、三苯基膦(纯度98%)购自上海达瑞精细化学品有限公司,(S)-叔亮氨醇(纯度99%)购自上海毕得医药科技有限公司,L-苯丙氨醇(纯度99%)购自上海书亚科技有限公司,L-色氨醇(纯度99%)购自阿达玛斯公司,1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDCI)(纯度98%)、1-羟基苯并三唑(HOBt) (纯度99%)、二环己基碳二亚胺(DCC)(纯度99%)、4-二甲氨基吡啶(DMAP)(纯度99%)购自安耐吉公司,二乙胺基三氟化硫(DAST)(纯度99%)购自百灵威科技有限公司,其它试剂均为分析纯。
1.2 实验方法
1.2.1 羟基酰胺3的制备(以化合物3a为例)
向35 mL反应管中加入2-(二苯基膦基)苯甲酸(0.14 mmol, 43 mg),(S)-叔亮氨醇(0.21 mmol, 25 mg),EDCI(0.42 mmol, 80 mg),HOBt(0.31 mmol, 42 mg),反应管用氮气置换3次后加入干燥的N, N-二甲基甲酰胺(DMF, 2 mL),用聚四氟乙烯旋塞将反应管封闭,25 ℃下搅拌24 h。向反应液中加入水(10 mL),乙酸乙酯(25 mL×3)萃取,合并有机相并用饱和氯化钠溶液洗涤,无水Na2SO4干燥,过滤,浓缩,之后以V(石油醚):V(丙酮)=5:1为洗脱液柱层析得到目标产物3a,产率98%。
化合物3a(R=t-Bu) 产率98%;mp 116~118 ℃; [α]20D -88.7(c 0.80, CHCl3); 1H NMR(CDCl3, 300 MHz), δ:7.69(ddd, J=7.6, 3.9, 1.5 Hz, 1H), 7.49~7.28(m, 10H), 7.23~7.25(m, 2H), 6.99(ddd, J=7.7, 4.3, 1.3 Hz, 1H), 5.66(d, J=9.8 Hz, 1H), 3.99~4.04(m, 1H), 3.83~3.68(m, 1H), 3.31~3.28(m, 1H), 2.93~2.90(m, 1H), 0.77(s, 9H); 13C NMR(CDCl3, 150 MHz), δ:26.5, 33.0, 60.3, 62.3, 128.2(d, J=6.2 Hz), 128.5(d, J=7.1 Hz), 128.9, 129.02, 129.04, 129.2(d, J=6.0 Hz), 130.2, 133.2(d, J=14.8 Hz), 133.4, 133.5(d, J=5.2 Hz), 133.6, 134.3, 135.3(d, J=7.6 Hz), 135.9(d, J=7.3 Hz), 142.3(d, J=28.8 Hz), 169.9。
化合物3b(R=Bn) 产率98%;mp 142~144 ℃; [α]20D -78.2(c 0.56, CHCl3); 1H NMR(CDCl3, 400 MHz), δ:7.47(dd, J=7.6, 3.8 Hz, 1H), 7.16~7.39(m, 15H), 7.09(d, J=7.3 Hz, 2H), 6.95(dd, J=7.5, 4.3 Hz, 1H), 5.94(d, J=7.5 Hz, 1H), 4.18~4.23(m, 1H), 3.68(dd, J=11.5, 3.3 Hz, 1H), 3.44(dd, J=11.5, 4.7 Hz, 1H), 2.67~2.79(m, 2H); 13C NMR(CDCl3, 150 MHz), δ:36.5, 53.5, 63.3, 126.4, 127.8(d, J=5.2 Hz), 128.4, 128.5(d, J=7.1 Hz), 128.7(d, J=7.1 Hz), 128.9, 129.0, 129.1, 130.1, 133.6, 133.7(d, J=5.8 Hz), 133.8, 133.9, 134.6(d, J=17.5 Hz), 136.0(d, J=8.8 Hz), 136.3(d, J=9.0 Hz), 137.4, 141.5(d, J=26.4 Hz), 169.2。
化合物3c(R=Ph) 产率92%;mp 148~150 ℃; [α]20D +51.0(c 0.41, CHCl3); 1H NMR(CDCl3, 400 MHz), δ:7.64~7.67(m, 1H), 7.43~7.19(m, 15H), 7.08~7.05(m, 2H), 6.98~6.95(m, 1H), 6.37(d, J=7.6 Hz, 1H), 5.14~5.12(m, 1H), 3.83~3.70(m, 2H); 13C NMR(CDCl3, 150 MHz), δ:56.5, 66.2, 126.7, 127.7, 128.2(d, J=5.0 Hz), 128.5(d, J=7.0 Hz), 128.6, 128.8(d, J=6.9 Hz), 128.94, 128.99, 129.0, 130.3, 133.6(d, J=6.6 Hz), 133.7(d, J=6.3 Hz), 134.1, 134.4(d, J=18.0 Hz), 135.8(d, J=8.8 Hz), 136.3(d, J=8.7 Hz), 138.3, 141.4(d, J=26.7 Hz), 169.2。
化合物3d(R=3-Indolymethyl) 产率99%;mp 102~104 ℃; [α]20D -55.0(c 1.0, CHCl3); 1H NMR(CDCl3, 600 MHz), δ:7.97(s, 1H), 7.57(d, J=7.9 Hz, 1H), 7.48(ddd, J=7.5, 3.8, 1.4 Hz, 1H), 7.36~7.26(m, 11H), 7.25~7.22(m, 2H), 7.19(ddd, J=8.2, 7.0, 1.1 Hz, 1H), 7.10(ddd, J=8.0, 7.0, 1.0 Hz, 1H), 6.95~6.93(m, 2H), 5.99(d, J=7.4 Hz, 1H), 4.35~4.33(m, 1H), 3.73~3.69(m, 1H), 3.50~3.48(m, 1H), 2.95~2.85(m, 2H), 2.62~2.59(m, 1H); 13C NMR(CDCl3, 150 MHz), δ:26.2, 52.8, 63.9, 111.1, 111.3, 118.6, 119.3, 121.9, 122.5, 127.4, 127.8(d, J=5.1 Hz), 128.5(d, J=7.1 Hz), 128.6(d, J=7.2 Hz), 128.8, 128.9, 130.1, 133.6(d, J=5.9 Hz), 133.7(d, J=6.0 Hz), 133.9, 134.7(d, J=18.2 Hz), 136.1, 136.2(d, J=9.2 Hz), 136.3(d, J=9.2 Hz), 141.5(d, J=26.2 Hz), 169.4; HRMS计算值C30H28N2O2P [M+H]+ 479.1883, 实测值479.1864。
1.2.2 由羟基酰胺制备膦-噁唑啉(PHOX)化合物4(以4a为例)
向干燥的35 mL反应管中加入羟基酰胺3a(0.05 mmol, 20 mg),PPh3(0.15 mmol, 39 mg),反应管用N2气置换3次后加入乙腈(0.5 mL)。0 ℃搅拌下依次滴加CCl4(0.5 mmol, 0.048 mL),NEt3(0.25 mmol, 0.035 mL)。TLC跟踪至底物完全转化。向反应液中加入水(10 mL),乙酸乙酯(25 mL×3)萃取,合并有机相并用饱和氯化钠溶液洗涤,无水Na2SO4干燥,过滤,浓缩,之后以二氯甲烷为洗脱液柱层析得到目标产物4a,产率88%。
化合物4a(R=t-Bu) 产率88%;mp 113~114 ℃(lit., 114~116 ℃)[8]; [α]20D -57.6(c 0.92, CHCl3)[lit., [α]20D -55.2(c 0.6, CHCl3)][8]; 1H NMR(CDCl3, 300 MHz), δ:7.96~7.94(m, 1H), 7.21~7.39(m, 12H), 6.85~6.88(m, 1H), 3.98~4.11(m, 2H), 3.87(dd, J=10.2, 8.4 Hz, 1H), 0.72(s, 9H); 13C NMR(CDCl3, 150 MHz), δ:25.6, 33.5, 68.1, 76.6, 127.8, 128.1, 128.20, 128.25, 128.35, 129.7, 130.2, 131.9(d, J=19.6 Hz), 133.4(d, J=20.2 Hz), 134.0, 134.2(d, J=20.5 Hz), 138.1(d, J=9.6 Hz), 138.4(d, J=12.3 Hz), 138.6(d, J=25.8 Hz), 162.5。
化合物4b(R=Bn) 产率70%;mp 117~119 ℃; [α]20D +26.3(c 1.14, CHCl3)[lit., [α]20D +25.8(c 1.10, CHCl3)][10]; 1H NMR(CDCl3, 600 MHz), δ:7.86(ddd, J=7.7, 3.5, 1.4 Hz, 1H), 7.36~7.17(m, 15H), 7.09~7.05(m, 2H), 6.86(ddd, J=7.8, 4.3, 1.3 Hz, 1H), 4.37~4.32(m, 1H), 4.03(t, J=8.8 Hz, 1H), 3.77(t, J=7.9 Hz, 1H), 2.92(dd, J=13.9, 5.1 Hz, 1H), 2.11(dd, J=13.9, 9.2 Hz, 1H); 13C NMR(CDCl3, 150 MHz), δ:41.0, 67.8, 71.3, 126.2, 127.8, 128.2, 128.30, 128.34, 128.39, 128.4, 128.6, 128.9, 129.8, 130.3, 131.4(d, J=18.2 Hz), 133.4, 133.7(d, J=20.9 Hz), 134.3(d, J=21.2 Hz), 137.1, 137.8(d, J=9.4 Hz), 138.1, 138.8(d, J=25.2 Hz), 163.7。
化合物4c(R=Ph) 产率85%;mp 113~115 ℃(lit., 118~119 ℃)[11]; [α]20D -26.7(c 1.45, CHCl3)[lit., [α]20D -28.1(c 1.43, CHCl3)][11]; 1H NMR(CDCl3, 400 MHz), δ:8.02~8.00(m, 1H), 7.40~7.28(m, 12H), 7.20~7.19(m, 3H), 6.92~6.89(m, 3H), 5.22(t, J=9.6 Hz, 1H), 4.56(dd, J=10.2, 8.2 Hz, 1H), 3.93(dd, J=9.2, 8.7 Hz, 1H); 13C NMR(CDCl3, 150 MHz), δ:70.0, 74.2, 126.5, 126.9, 127.9, 128.2, 128.30, 128.36, 128.4, 128.5, 130.2, 130.5, 131.3(d, J=19.0 Hz), 133.5, 133.7, 133.8, 134.1(d, J=21.0 Hz), 137.6(d, J=10.2 Hz), 137.8(d, J=12.1 Hz), 138.9(d, J=25.0 Hz), 141.9, 164.5。
化合物4d(R=3-Indolymethyl) 产率65%;mp 90~92 ℃; [α]20D +19.1(c 2.35, EtOH) [lit., [α]20D +17.9(c 2.35, EtOH)][11]; 1H NMR(CDCl3, 400 MHz), δ:7.96(s, 1H), 7.88(ddd, J=7.7, 3.6, 1.5 Hz, 1H), 7.55(d, J=7.8 Hz, 1H), 7.38~7.27(m, 13H), 7.19~7.10(m, 2H), 6.88~6.85(m, 2H), 4.50~4.44(m, 1H), 4.05(t, J=8.8 Hz, 1H), 3.81(t, J=7.9 Hz, 1H), 3.01(dd, J=14.6, 5.0 Hz, 1H), 2.30(dd, J=14.7, 9.3 Hz, 1H); 13C NMR(CDCl3, 150 MHz), δ:30.6, 66.9, 71.9, 110.9, 112.3, 118.6, 119.2, 121.9, 122.0, 127.4, 128.2, 128.31(d, J=3.8 Hz), 128.39 128.4, 128.6, 129.8, 130.3, 131.5(d, J=18.2 Hz), 133.4, 133.8(d, J=20.7 Hz), 134.2(d, J=21.2 Hz), 136.0, 137.79(d, J=18.0 Hz), 137.84(d, J=3.0 Hz), 138.7(d, J=25.3 Hz), 163.6。
1.2.3 不对称烯丙基化反应合成化合物6
向干燥的35 mL反应管中加入Pd2(dba)3(0.015 mmol, 13.7 mg),(S)-t-BuPHOX(4a, 0.03 mmol, 11.6 mg),反应管用氮气置换3次后加入乙醚(5 mL),20 ℃搅拌30 min。将β-酮酯5溶于乙醚(10 mL)加入至反应体系中,20 ℃搅拌反应。TLC跟踪至底物完全转化。过滤,浓缩,之后以V(石油醚):V(乙醚)=20:1为洗脱液柱层析得到目标产物6,产率80%,84% e.e.。
1H NMR(CDCl3, 300 MHz), δ:5.77~5.63(m, 1H), 5.09~4.02(m, 2H), 2.34~2.13(m, 4H), 1.97~1.82(m, 3H), 1.74~1.65(m, 1H), 1.01(s, 3H); 13C NMR(CDCl3, 100 MHz), δ:18.2, 21.4, 34.6, 37.2, 40.5, 47.7, 117.7, 133.4, 222.5;HPLC:ChiralPak AS-H, n-hexane/i-PrOH=95/5, flow rate=1 mL/min, tr(minor)=4.57 min, tr(major)=5.07 min。
2 结果与讨论
2.1 2-(二苯基膦基)苯甲酸与手性氨基醇缩合反应条件的筛选
以2-(二苯基膦基)苯甲酸1a和(S)-叔亮氨醇2a为起始原料对缩合反应进行了条件优化(表 1)。开始,我们尝试将羧酸转化为酰氯[13-14]或混酐之后与氨基醇反应以求合成酰胺,但结果并不理想(表 1, Entries 1~2)。接下来考察了一些常用的缩合条件。当采用DCC/DMAP/CH2Cl2反应体系时[15], 室温条件下便可以获得30%的收率;提高温度至50 ℃,产率只有小幅提升(表 1, Entries 4, 5)。如果用HOBt替换DMAP[16]则可以获得约65%的收率(表 1, Entry 6)。随后对溶剂的简单筛选表明DMF是最优的反应介质(表 1, Entries 7~9)。同时,(S)-叔亮氨醇的用量也可以降至1.5倍化学计量而并未引起产率明显降低(表 1, Entry 10)。但使用DCC存在反应副产物与目标物不易分离的问题,当改用EDCI[17]时,分离更加方便且收率提高到98%(表 1, Entry 11)。综上所述,最佳反应条件为2-(二苯基膦基)苯甲酸(0.1 mmol),(S)-叔亮氨醇(0.15 mmol),EDCI(0.3 mmol),HOBt(0.22 mmol),在DMF中室温下反应。
 表 1
缩合反应的条件优化
Table 1.
Optimization of the condensation reaction conditions
表 1
缩合反应的条件优化
Table 1.
Optimization of the condensation reaction conditions

Entry Reagents(mmol) Solvent Temperature/℃ Yield/%a 1 (COCl)2(0.2), DMF(0.02) CH2Cl2 25 - 2 MeOC(O)Cl(0.12), NMM(0.12) THF 0 - 3 CDI(0.3), DMAP(0.1) CH2Cl2 25 20 4 DCC(0.3), DMAP(0.22) CH2Cl2 25 30 5 DCC(0.3), DMAP(0.22) CH2Cl2 50 35 6 DCC(0.3), HOBt(0.22) CH2Cl2 50 65 7 DCC(0.3), HOBt(0.22) CH2Cl2 25 64 8 DCC(0.3), HOBt(0.22) THF 25 75 9 DCC(0.3), HOBt(0.22) DMF 25 80 10b DCC(0.3), HOBt(0.22) DMF 25 78 11b EDCI(0.3), HOBt(0.22) DMF 25 98 a.isolated yield; b.2a(0.15 mmol). DMF=N, N-dimethylformamide, THF=tetrahydrofuran, NMM=N-methyl morphofine, CDI=1, 1′-carbonyldiimidazole, DMAP=4-dimethylaminopyridine, DCC=dicyclohexylcarbodiimide, HOBt=1-hydroxybenzotriazole, EDCI=1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride. 2.2 由羟基酰胺关环合成膦-噁唑啉(PHOX)化合物的反应条件的筛选
以化合物3a为底物考察了不同的关环条件(表 2)。通过磺酰基(Ts-或Ms-)活化羟基酰胺,碱性条件下发生亲核取代是合成噁唑啉环的常用方法。但此处采用TsCl/NEt3及MsCl/NEt3反应体系均未得到目标物(表 2, Entries 1, 2)。有文献[18]报道DAST亦可用于合成噁唑啉,于是,我们也使用DAST为反应试剂,对其用量及反应时间进行了探索,但最后仅能获得40%的收率(表 2, Entries 3~8)。参考有关文献[19-21],我们接下来采用PPh3/CCl4/NEt3的反应体系,对反应时间及温度进行了简单考察(表 2, Entries 9~11),结果表明,0 ℃条件下反应2 h为宜。最终确立的最佳反应条件为羟基酰胺(0.05 mmol),PPh3(0.15 mmol),NEt3(0.25 mmol),CCl4(0.50 mmol),MeCN为溶剂,0 ℃条件下反应,目标物4a的收率可达88%。
表 2
噁唑啉环形成反应的条件优化
Table 2.
Optimization of the oxazoline ring formation reaction conditions

Entry Reagents(mmol) Solvent Time Temperature/℃ Yield/%a 1 TsCl(0.10), Et3N(0.25) CH2Cl2 12 h 50 - 2 MsCl(0.10), Et3N(0.50), DMAP(0.0025) CH2Cl2 30 min 0~25 - 3 DAST(0.075) CH2Cl2 12 h -78 - 4 DAST(0.075) CH2Cl2 4 h -78 10 5 DAST(0.075) CH2Cl2 2 h -78 20 6 DAST(0.06) CH2Cl2 2 h -78 25 7 DAST(0.06) CH2Cl2 1 h -78 30 8 DAST(0.06) CH2Cl2 5 min -78 40 9 PPh3(0.15), Et3N(0.25), CCl4(0.50) MeCN 4 h 25 20 10 PPh3(0.15), Et3N(0.25), CCl4(0.50) MeCN 1 h 25 50 11 PPh3(0.15), Et3N(0.25), CCl4(0.50) MeCN 2 h 0 88 a.isolated yield. Ts=tosyl, Ms=methanesulfonyl, DMAP=4-dimethylaminopyridine, DAST=diethylaminosulfur trifluoride 2.3 膦-噁唑啉(PHOX)化合物的合成
在最优的反应条件下,以带有不同取代基的手性氨基醇为起始原料,考察了新合成方法的底物适用性(表 3)。实验结果表明,2-(二苯基膦基)苯甲酸与不同的氨基醇的缩合反应均可以高效生成羟基酰胺类化合物;接下来的关环反应亦可以顺利进行,最终以高的总收率得到一系列膦-噁唑啉(PHOX)化合物。
表 3
膦-噁唑啉(PHOX)化合物的合成
Table 3.
Synthesis of phosphinooxazolines(PHOX ligands)


2.4 化合物4a用于不对称催化反应
接下来,我们将合成的配体应用于钯催化的不对称催化反应。以化合物4a为配体,经钯催化β-酮酯的脱羧Tsuji烯丙基化反应,可以完成极具挑战性的季碳手性中心的构建,反应的收率及对映选择性与文献[22]所报道的结果相当。
3 结论
本文报道了一种合成膦-噁唑啉(PHOX)化合物的简便方法。以商品化的2-(二苯基膦基)苯甲酸和手性氨基醇为起始原料,在1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐/1-羟基苯并三唑(EDCI/HOBt)反应体系下发生缩合得到羟基酰胺类化合物。随后将羟基酰胺置于三苯基膦/三乙胺/四氯化碳/乙腈(PPh3/NEt3/CCl4/MeCN)反应体系中便可以实现噁唑啉环的高效构建。与以往的合成路线相比,该方法具有原料简单易得,反应条件温和,操作简单等特点,通过选择不同反应底物可以实现噁唑啉环上不同取代基的灵活引入,因此,该反应为膦-噁唑啉(PHOX)化合物的合成提供了简便的新方法。
-
-
[1]
Sprinz J, Helmchen G. Phosphinoaryl-and Phosphinoalkyl Oxazolines as New Chiral Ligands for Enantioselective Catalysis:Very High Enantioselectivity in Palladium Catalyzed Allylic Substitutions[J]. Tetrahedron Lett, 1993, 34(11): 1769-1772. doi: 10.1016/S0040-4039(00)60774-8
-
[2]
von Matt P, Pfaltz A. Chiral Phosphinoaryldihydrooxazoles as Ligands in Asymmetric Catalysis:Pd-Catalyzed Allylic Substitution[J]. Angew Chem Int Ed, 1993, 32(4): 566-568. doi: 10.1002/(ISSN)1521-3773
-
[3]
Dawson G J, Frost C, Williams J M J. Asymmetric Palladium Catalysed Allylic Substitution Using Phosphorus Containing Oxazoline Ligands[J]. Tetrahedron Lett, 1993, 34(19): 3149-3150. doi: 10.1016/S0040-4039(00)93403-8
-
[4]
Helmchen G, Pfaltz A. Phosphinooxazolines——A New Class of Versatile, Modular P, N-Ligands for Asymmetric Catalysis[J]. Acc Chem Res, 2000, 33(6): 336-345. doi: 10.1021/ar9900865
-
[5]
Bausch C C, Pfaltz A. PHOX Ligands. Privileged Chiral Ligands and Catalysts[M]. Wiley-VCH Verlag GmbH & Co. KgaA, 2006:221-256.
-
[6]
Hargaden G C, Guiry P J. Recent Applications of Oxazoline-Containing Ligands in Asymmetric Catalysis[J]. Chem Rev, 2009, 109(6): 2505-2550. doi: 10.1021/cr800400z
-
[7]
Carroll M P, Guiry P J. P, N Ligands in Asymmetric Catalysis[J]. Chem Soc Rev, 2014, 43(3): 819-833. doi: 10.1039/C3CS60302D
-
[8]
Allen J V, Dawson G J, Frost C G. Preparation of Novel Sulfur and Phosphorus Containing Oxazolines as Ligands for Asymmetric Catalysis[J]. Tetrahedron, 1994, 50(3): 799-808. doi: 10.1016/S0040-4020(01)80795-X
-
[9]
Gant T G, Meyers A I. The Chemistry of 2-Oxazolines(1985 present)[J]. Tetrahedron, 1994, 50(8): 2297-2360. doi: 10.1016/S0040-4020(01)86953-2
-
[10]
Koch G, Lloyd-jones G C, Loiseleur O. Synthesis of Chiral (Phosphinoaryl) Oxazolines, a Versatile Class of Ligands for Asymmetric Catalysis[J]. Recl Trav Chim Pays-Bas, 1995, 114(4/5): 206-210.
-
[11]
Peer M, de Jong J C, Kiefer M. Preparation of Chiral Phosphorus, Sulfur and Selenium Containing 2-Aryloxazolines[J]. Tetrahedron, 1996, 52(21): 7547-7583. doi: 10.1016/0040-4020(96)00267-0
-
[12]
Tani K, Behenna D C, McFadden R M. A Facile and Modular Synthesis of Phosphinooxazoline Ligands[J]. Org Lett, 2007, 9(13): 2529-2531.
-
[13]
Sedinkin S L, Rath N P, Bauer E B. Synthesis and Structural Characterization of New Phosphinooxazoline Complexes of Iron[J]. J Organomet Chem, 2008, 693(18): 3081-3091. doi: 10.1016/j.jorganchem.2008.06.031
-
[14]
Wu W Q, Peng Q, Dong D X. A Dramatic Switch of Enantioselectivity in Asymmetric Heck Reaction by Benzylic Substituents of Ligands[J]. J Am Chem Soc, 2008, 130(30): 9717-9725. doi: 10.1021/ja7104174
-
[15]
Marinho V R, Rodrigues A I, Burke A J. Novel Chiral P, O-Ligands for Homogeneous Pd(0) Catalysed Asymmetric Allylic Alkylation Reactions[J]. Tetrahedron:Asymmetry, 2008, 19(4): 454-458. doi: 10.1016/j.tetasy.2008.01.024
-
[16]
Zhu S F, Xie J B, Zhang Y Z. Well-Defined Chiral Spiro Iridium/Phosphine-Oxazoline Cationic Complexes for Highly Enantioselective Hydrogenation of Imines at Ambient Pressure[J]. J Am Chem Soc, 2006, 128(39): 12886-12891. doi: 10.1021/ja063444p
-
[17]
Zhang X P, Liang H Y, Cao Z K. Enantioselective Synthesis of Axially Chiral Biaryl Monophosphine Oxides via Direct Asymmetric Suzuki Coupling and DFT Investigations of the Enantioselectivity[J]. ACS Catal, 2014, 4(5): 1390-1397. doi: 10.1021/cs500208n
-
[18]
Burrell G, Evans J M, Jones G E. The Action of Diethylaminosulphur Trifluoride(dast) on Trans-4-Amido-3-chromanols:Preparation of cis-Amidoalcohols via Oxazolines[J]. Tetrahedron Lett, 1990, 31(25): 3649-3652. doi: 10.1016/S0040-4039(00)94467-8
-
[19]
Vorbrüggen H, Krolikiewicz K. A Simple Synthesis of Δ2-Oxazolines, Δ2-Oxazines, Δ2-Thiazolines and Δ2-Imidazolines[J]. Tetrahedron Lett, 1981, 22(45): 4471-4474. doi: 10.1016/S0040-4039(01)93017-5
-
[20]
Meyers A I, Hoyer D. Stereochemistry of the Ph3P-CCl4 Mediated Cyclization of Carboxylic Acids and 1, 2-Amino Alcohols(Vorbruggen Method)[J]. Tetrahedron Lett, 1985, 26(39): 4687-4690. doi: 10.1016/S0040-4039(00)94924-4
-
[21]
Vorbrüggen H, Krolikiewicz K. A Simple Synthesis of Δ2-Oxazines, Δ2-Oxazines, Δ2-Thiazolines and 2-Substituted Benzoxazoles[J]. Tetrahedron, 1993, 49(41): 9353-9372. doi: 10.1016/0040-4020(93)80021-K
-
[22]
Robert A Craig Ⅱ, Steven A Loskot, Brian M Stoltz. Palladium-Catalyzed Enantioselective Decarboxylative Allylic Alkylation of Cyclopentanones[J]. Org Lett, 2015, 17(21): 5160-5163. doi: 10.1021/acs.orglett.5b02376
-
[1]
-
表 1 缩合反应的条件优化
Table 1. Optimization of the condensation reaction conditions
Entry Reagents(mmol) Solvent Temperature/℃ Yield/%a 1 (COCl)2(0.2), DMF(0.02) CH2Cl2 25 - 2 MeOC(O)Cl(0.12), NMM(0.12) THF 0 - 3 CDI(0.3), DMAP(0.1) CH2Cl2 25 20 4 DCC(0.3), DMAP(0.22) CH2Cl2 25 30 5 DCC(0.3), DMAP(0.22) CH2Cl2 50 35 6 DCC(0.3), HOBt(0.22) CH2Cl2 50 65 7 DCC(0.3), HOBt(0.22) CH2Cl2 25 64 8 DCC(0.3), HOBt(0.22) THF 25 75 9 DCC(0.3), HOBt(0.22) DMF 25 80 10b DCC(0.3), HOBt(0.22) DMF 25 78 11b EDCI(0.3), HOBt(0.22) DMF 25 98 a.isolated yield; b.2a(0.15 mmol). DMF=N, N-dimethylformamide, THF=tetrahydrofuran, NMM=N-methyl morphofine, CDI=1, 1′-carbonyldiimidazole, DMAP=4-dimethylaminopyridine, DCC=dicyclohexylcarbodiimide, HOBt=1-hydroxybenzotriazole, EDCI=1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride.  下载: 导出CSV
下载: 导出CSV
表 2 噁唑啉环形成反应的条件优化
Table 2. Optimization of the oxazoline ring formation reaction conditions
Entry Reagents(mmol) Solvent Time Temperature/℃ Yield/%a 1 TsCl(0.10), Et3N(0.25) CH2Cl2 12 h 50 - 2 MsCl(0.10), Et3N(0.50), DMAP(0.0025) CH2Cl2 30 min 0~25 - 3 DAST(0.075) CH2Cl2 12 h -78 - 4 DAST(0.075) CH2Cl2 4 h -78 10 5 DAST(0.075) CH2Cl2 2 h -78 20 6 DAST(0.06) CH2Cl2 2 h -78 25 7 DAST(0.06) CH2Cl2 1 h -78 30 8 DAST(0.06) CH2Cl2 5 min -78 40 9 PPh3(0.15), Et3N(0.25), CCl4(0.50) MeCN 4 h 25 20 10 PPh3(0.15), Et3N(0.25), CCl4(0.50) MeCN 1 h 25 50 11 PPh3(0.15), Et3N(0.25), CCl4(0.50) MeCN 2 h 0 88 a.isolated yield. Ts=tosyl, Ms=methanesulfonyl, DMAP=4-dimethylaminopyridine, DAST=diethylaminosulfur trifluoride
下载: 导出CSV
-



 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 2
- 文章访问数: 1502
- HTML全文浏览量: 118
 下载:
下载:
