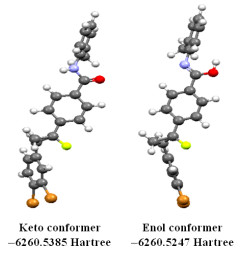
Figure 1.
Possible optimized structural conformers of DBFB
X-ray Structure and Density Functional Theory Investigations of 4-((2R)-2-(3, 4-dibromophenyl)-1-fluoro cyclopropyl)-N-(o-tolyl)benzamide Compound
Yamina BENELHADJ-DJELLOUL , Nourdine BOUKABCHA , Nadia BENHALIMA , Salem YAHIAOUI , Abdelkader CHOUAIH , Abdelouahab ZANOUN

In the past few decades, pyrethroids have a large spectrum of insecticidal activity. However, synthetic ones exhibit higher photostability and are more effective for practical use. They have a higher insecticidal capacity and especially a low toxicity for the mammals. The pyrethroids structures obtained by X-ray diffraction data and their structural requirements for insecticidal activity have been investigated by several authors[1-4]. Among them, carboxylic esters containing cyclopropane ring in their structure have powerful insecticidal activity[5-8]. In addition, the cyclopropane ring is a main structural part in many synthetic and natural compounds that exhibit a wide range of biological activities from enzyme inhibition to antibiotic, herbicidal, antitumor, and HIV antiviral activities as non-nucleoside reverse transcriptase inhibitors[9-11]. It is also known that the cyclopropane ring can be a crucial feature for the presence of asymmetric carbons as is found in the structures of several pyrethroids[12]. The study of the conformational behavior of pyrethriods is extremely important. The structural, conformational and physicochemical properties of these compounds may give information about the mechanism of their biological activity[13, 14]. This activity is related to molecular structure and strongly depends on the stereochemistry at the asymmetric centers. On the other hand, theoretical calculations were used to further study the structure-activity relationship of several heterocyclic compounds[15-17].
In this paper, we propose a comparative study between the experimental X-ray diffraction structure and the optimized geometry predicted from ab initio molecular orbital calculations using the Becke-Lee-Yang-Part's three-parameter hybrid functional (B3LYP) and HF methods at 6-31G(d, p) basis set performed on DBFB molecule. The investigated compound, 4-((2R)-2-(3, 4-dibromophenyl)-1-fluorocyclopropyl)-N-(o-tolyl)benzamide (DBFB), appears as a useful intermediate in the synthesis of some pyrethroid insecticides. This compound was kindly supplied by the French company RUSSEL UCLAF. With the goal of understanding the nature and reactivity of the molecule, some reactivity descriptors such as ionization energy, electron affinity, HOMO-LUMO energy gap, chemical potential, molecular softness, hardness and electrophilicity index have been calculated using the DFT/ B3LYP/6-31G(d, p) level of theory.
A single crystal of the title compound in appropriate dimensions was selected for X-ray diffraction measurements performed on a Philips Enraf Nonius (four-circle diffractometer) with CCD area detector using graphite-monochromated MoKα radiation (λ = 0.71073 Å). The data were corrected for Lorentz and Polarization effects. The X-ray data were collected at 298 K. A total of 3053 reflections were collected in the range of 2 < θ < 29.6° by using an ω scan mode, of which 1464 observed reflections with I > 2σ(I) were used. The structure was solved by direct methods implemented in SHELXS2013[18]. A Fourier synthesis revealed the complete structure, which was refined by full-matrix least-squares. All non-H atoms were refined anisotropically. The H atoms were located from a difference Fourier map and included in the refinement with the isotropic temperature factor of the carrier atom. The final least-squares cycle using SHELXL gave R = 0.0639 for all reflections with I > 2σ(I), wR = 0.222, S = 1.51, (Δρ)min = −0.94 and (Δρ)max = 1.45 e/Å3.
In order to explain the biological activity of the compound, we used theoretical methods from quantum chemistry. Ab initio geometry optimization on C23H18Br2FNO was performed starting from the experimental data refinement (X-ray data). Geometry optimizations and harmonic wavenumbers for the normal modes of vibration were calculated using the Density Functional Theory, Becke's three parameter hybrid functional using the LYP correlation Functional (B3LYP) and HF theory with the 6-31G (d, p) basis set[19-21]. The compute vibrational frequencies were attributed by means of the potential energy distribution (PED) investigation of all fundamental vibration modes using VEDA 4 program[22]. Such combination is being used with good results for organic molecules[23] and hydrogen-bonded systems[24, 25] and represents a good compromise between economy of computational resources, accuracy and applicability to manyatoms molecules. All calculations were performed with the GAUSSIAN 09 program[26]. Other molecular properties of DBFB were evaluated using the same level of theory.
To determine the tautomeric forms of DBFB molecule as the initial point for further calculations, the molecule was submitted to a rigorous conformation analysis. The tautomerism of organic compounds has been studied by theoretical calculations through several quantum mechanics approaches[27, 28]. In this study, the Gaussian 09 software was used to perform the conformational analysis. The two possible conformations of DBFB are shown in Fig. 1. The stability analysis as obtained from energy minimization shows that the keto conformer (−6260.5385 Hartree) is the most stable compared to the enol conformer (−6260.5247 Hartree). The keto form predominates at equilibrium for most ketones. Therefore, our study has focused on this particular form of DBFB.
The most stable geometry and other possible conformations of the compound have been determined from the potential energy surface (PES) using B3LYP/6-31G** method. During the analysis all geometrical parameters are simultaneously relaxed while the N(1)−C(16)−C(13)−C(14) dihedral angle varies in a step of 10o from 0o to 360o[29]. The potential energy profile which reflects the stability of the possible conformers of the molecule is shown in Fig. 2.
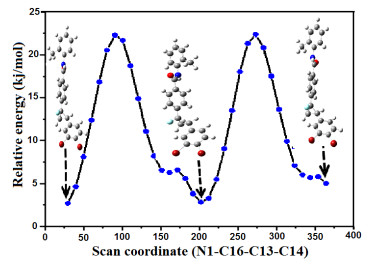
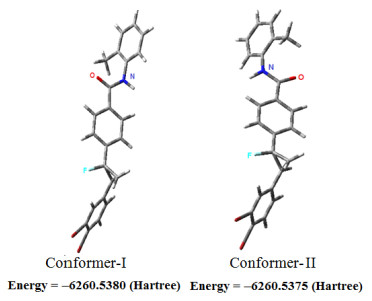
Two different conformers I and II have been determined for DBFB by PES analysis and are shown in Fig. 3. The rotation about C(13)−C(16) single bond produces two conformers: conformer I = trans and conformer II = cis. In the most stable geometry, the CH3 group is on the side of oxygen atom. In order to provide the accurate structural parameters of the compound, the most stable conformer is optimized with the B3LYP/6-31G** method.
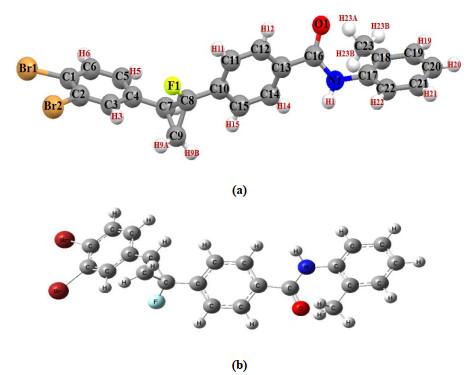
The X-ray structure of the molecule with atomic labeling and optimized geometry is shown in Fig. 4. The experimental geometrical parameters of DBFB (bonds lengths, bond angles and dihedral angles) are listed in Table 1, in which the corresponding optimized geometrical parameters obtained by Hartree-Fock and DFT (B3LYP) levels of theory using 6-31G (d, p) basis set are also given.
 DownLoad:
CSV
DownLoad:
CSV
| Bond | Dist. (Å) | Bond | Dist. (Å) | |||||
| HF | B3LYP | X-ray | HF | B3LYP | X-ray | |||
| Br(1)–C(1) | 1.897 | 1.900 | 1.854(12) | C(17)–C(22) | 1.386 | 1.400 | 1.40(3) | |
| Br(2)–C(2) | 1.898 | 1.903 | 1.858(15) | C(17)–C(18) | 1.395 | 1.409 | 1.36(2) | |
| N(1)–C(16) | 1.354 | 1.383 | 1.315(13) | F(1)–C(8) | 1.360 | 1.381 | 1.36(2) | |
| N(1)–C(17) | 1.427 | 1.426 | 1.410(13) | C(13)–C(14) | 1.390 | 1.400 | 1.336(17) | |
| N(1)–H(1) | 0.994 | 1.010 | 0.8600 | C(7)–C(9) | 1.508 | 1.520 | 1.48(2) | |
| C(16)–O(1) | 1.202 | 1.224 | 1.224(13) | C(7)–C(8) | 1.508 | 1.531 | 1.501(19) | |
| C(16)–C(13) | 1.501 | 1.503 | 1.467(13) | C(14)–C(15) | 1.382 | 1.391 | 1.373(16) | |
| C(4)–C(3) | 1.387 | 1.401 | 1.371(19) | C(1)–C(6) | 1.385 | 1.395 | 1.30(3) | |
| C(4)–C(5) | 1.390 | 1.402 | 1.38(2) | C(1)–C(2) | 1.385 | 1.398 | 1.41(2) | |
| C(4)–C(7) | 1.496 | 1.490 | 1.483(13) | C(2)–C(3) | 1.385 | 1.394 | 1.365(17) | |
| C(12)–C(11) | 1.389 | 1.392 | 1.382(16) | C(10)–C(15) | 1.391 | 1.402 | 1.382(19) | |
| C(12)–C(13) | 1.387 | 1.402 | 1.41(2) | C(10)–C(11) | 1.390 | 1.402 | 1.37(2) | |
| C(19)–C(20) | 1.386 | 1.394 | 1.40(3) | C(10)–C(8) | 1.498 | 1.490 | 1.468(15) | |
| C(19)–C(18) | 1.390 | 1.401 | 1.414(18) | C(8)–C(9) | 1.485 | 1.496 | 1.48(2) | |
| C(21)–C(22) | 1.385 | 1.393 | 1.37(2) | C(18)–C(23) | 1.509 | 1.506 | 1.48(3) | |
| C(21)–C(20) | 1.389 | 1.394 | 1.37(3) | C(6)–C(5) | 1.380 | 1.391 | 1.39(2) | |
| Bond angle (°) | HF | B3LYP | X-ray | Bond angle (°) | HF | B3LYP | X-ray | |
| C(16)–N(1)–C(17) | 123.32 | 124.67 | 125.2(9) | C(3)–C(2)–C(1) | 120.26 | 120.34 | 118.0(14) | |
| C(17)–N(1)–H(1) | 116.41 | 115.34 | 117.4(10) | C(3)–C(2)–Br(2) | 117.25 | 117.49 | 118.5(12) | |
| O(1)–C(16)–N(1) | 122.83 | 122.85 | 122.5(9) | C(1)–C(2)–Br(2) | 122.42 | 122.15 | 123.5(11) | |
| O(1)–C(16)–C(13) | 121.05 | 121.53 | 121.2(9) | C(4)–C(3)–C(2) | 118.21 | 120.90 | 123.6(14) | |
| N(1)–C(16)–C(13) | 116.10 | 115.60 | 116.3(9) | C(15)–C(10)–C(11) | 118.21 | 118.71 | 118.9(11) | |
| C(3)–C(4)–C(5) | 120.35 | 118.13 | 116.6(12) | C(15)–C(10)–C(8) | 119.99 | 120.00 | 120.8(13) | |
| C(3)–C(4)–C(7) | 123.30 | 123.12 | 126.0(12) | C(11)–C(10)–C(8) | 121.18 | 121.11 | 120.3(12) | |
| C(5)–C(4)–C(7) | 118.48 | 118.73 | 117.4(13) | C(14)–C(15)–C(10) | 120.42 | 120.51 | 119.7(13) | |
| C(11)–C(12)–C(13) | 120.54 | 120.63 | 119.5(13) | C(14)–C(15)–H(15) | 119.51 | 120.18 | 120.1 | |
| C(20)–C(19)–C(18) | 121.65 | 121.99 | 121.2(18) | C(10)–C(11)–C(12) | 120.62 | 120.65 | 120.6(13) | |
| C(22)–C(21)–C(20) | 119.34 | 119.44 | 116.9(18) | F(1)–C(8)–C(10) | 111.73 | 112.09 | 112.7(12) | |
| C(22)–C(17)–N(1) | 118.58 | 117.94 | 118.8(15) | F(1)–C(8)–C(9) | 113.89 | 114.28 | 112.3(13) | |
| C(22)–C(17)–C(18) | 121.73 | 120.81 | 120.2(14) | C(10)–C(8)–C(9) | 124.81 | 124.60 | 124.4(13) | |
| N(1)–C(17)–C(18) | 120.60 | 121.63 | 120.9(14) | F(1)–C(8)–C(7) | 114.44 | 114.51 | 114.3(9) | |
| C(14)–C(13)–C(12) | 118.78 | 118.60 | 118.5(10) | C(10)–C(8)–C(7) | 122.57 | 121.72 | 123.8(12) | |
| C(14)–C(13)–C(16) | 118.01 | 117.62 | 121.0(11) | C(9)–C(8)–C(7) | 58.97 | 58.66 | 59.5(12) | |
| C(12)–C(13)–C(16) | 123.08 | 123.68 | 120.4(11) | C(19)–C(20)–C(21) | 119.86 | 119.71 | 121.1(14) | |
| C(9)–C(7)–C(4) | 124.54 | 123.33 | 120.1(12) | C(17)–C(18)–C(19) | 117.75 | 117.52 | 117.0(17) | |
| C(9)–C(7)–C(8) | 58.97 | 58.66 | 59.6(12) | C(17)–C(18)–C(23) | 121.73 | 122.19 | 124.3(13) | |
| C(4)–C(7)–C(8) | 121.98 | 122.97 | 121.8(11) | C(19)–C(18)–C(23) | 120.52 | 120.40 | 118.7(16) | |
| C(13)–C(14)–C(15) | 120.78 | 120.89 | 122.3(12) | C(1)–C(6)–C(5) | 120.97 | 120.30 | 123.8(17) | |
| C(6)–C(1)–C(2) | 119.13 | 119.15 | 118.3(13) | C(21)–C(22)–C(17) | 120.59 | 120.75 | 123.4(19) | |
| C(6)–C(1)–Br(1) | 117.87 | 118.75 | 122.5(13) | C(4)–C(5)–C(6) | 121.06 | 121.15 | 119.3(17) | |
| C(2)–C(1)–Br(1) | 122.99 | 122.08 | 119.2(14) | C(7)–C(9)–C(8) | 60.48 | 61.11 | 60.8(8) | |
| Dihedral angle (°) | HF | B3LYP | X-ray | Dihedral angle (°) | HF | B3LYP | X-ray | |
| C(17)–N(1)–C(16)–C(13) | 176.80 | 175.64 | –176.6(15) | C(15)–C(10)–C(8)–F(1) | 8.47 | 7.94 | –13.1(17) | |
| C(17)–N(1)–C(16)–O(1) | –3.37 | –4.14 | 2(2) | C(11)–C(10)–C(8)–F(1) | –172.55 | –173.02 | 166.1(12) | |
| C(16)–N(1)–C(17)–C(22) | –111.92 | –121.09 | 118.5(17) | C(15)–C(10)–C(8)–C(9) | 152.26 | 152.54 | −154.7(18) | |
| C(16)–N(1)–C(17)–C(18) | 70.73 | 62.16 | –66(2) | C(11)–C(10)–C(8)–C(9) | –28.68 | –28.22 | 25(2) | |
| C(11)–C(12)–C(13)–C(14) | –1.08 | 1.06 | –6(2) | C(15)–C(10)–C(8)–C(7) | –133.32 | –133.87 | 131.6(15) | |
| C(11)–C(12)–C(13)–C(16) | –179.64 | 179.06 | 178.6(12) | C(11)–C(10)–C(8)–C(7) | 45.90 | 45.17 | –49.1(16) | |
| O(1)–C(16)–C(13)–C(14) | 24.93 | 22.47 | –34.4(18) | C(9)–C(7)–C(8)–F(1) | –105.64 | 106.26 | –102.5(14) | |
| N(1)–C(16)–C(13)–C(14) | –154.85 | –157.60 | 144.3(13) | C(4)–C(7)–C(8)–F(1) | –7.59 | –6.60 | 6.2(16) | |
| O(1)–C(16)–C(13)–C(12) | –153.17 | –156.24 | 140.9(13) | C(9)–C(7)–C(8)–C(10) | 111.11 | –113.93 | 113.2(17) | |
| N(1)–C(16)–C(13)–C(12) | 26.22 | 23.70 | –40.4(17) | C(4)–C(7)–C(8)–C(10) | 131.48 | 134.20 | –138.0(13) | |
| C(3)–C(4)–C(7)–C(9) | –13.27 | –30.20 | 16(2) | C(4)–C(7)–C(8)–C(9) | –112.49 | –111.87 | 108.7(15) | |
| C(5)–C(4)–C(7)–C(9) | 166.77 | 149.42 | –161.5(17) | N(1)–C(17)–C(18)–C(19) | 178.88 | 178.30 | 179.2(12) | |
| C(3)–C(4)–C(7)–C(8) | 58.80 | 41.50 | –55.2(16) | C(22)–C(17)–C(18)–C(23) | –179.01 | –178.38 | 174.3(19) | |
| C(5)–C(4)–C(7)–C(8) | –121.15 | –138.83 | 127.7(15) | C(20)–C(19)–C(18)–C(23) | 178.85 | 177.54 | –176.3(17) | |
| C(12)–C(13)–C(14)–C(15) | –1.08 | –1.42 | 7(2) | C(2)–C(1)–C(6)–C(5) | 0.09 | –0.11 | 8(3) | |
| C(16)–C(13)–C(14)–C(15) | –179.64 | 179.79 | –177.2(13) | Br(1)–C(1)–C(6)–C(5) | 179.95 | 179.88 | –174.7(16) | |
| Br(1)–C(1)–C(2)–C(3) | –179.82 | –179.85 | 175.2(10) | C(20)–C(21)–C(22)–C(17) | –0.58 | –0.57 | –3(3) | |
| C(6)–C(1)–C(2)–Br(2) | 179.80 | –179.87 | 174.0(15) | N(1)–C(17)–C(22)–C(21) | –178.20 | –177.61 | –179.0(18) | |
| Br(1)–C(1)–C(2)–Br(2) | 0.08 | 0.16 | –3.5(17) | C(18)–C(17)–C(22)–C(21) | –0.15 | –0.77 | 5(3) | |
| C(7)–C(4)–C(3)–C(2) | –179.77 | 179.27 | –176.2(12) | C(3)–C(4)–C(5)–C(6) | –0.06 | 0.39 | –1(2) | |
| C(1)–C(2)–C(3)–C(4) | –0.17 | 0.12 | 3(2) | C(7)–C(4)–C(5)–C(6) | 179.90 | –179.25 | 176.6(16) | |
| Br(2)–C(2)–C(3)–C(4) | 179.97 | –179.80 | –178.2(11) | C(4)–C(7)–C(9)–C(8) | 109.66 | –111.86 | –111.4(13) | |
| C(8)–C(10)–C(15)–C(14) | 179.08 | 0.984 | –179.1(13) | F(1)–C(8)–C(9)–C(7) | –105.64 | –105.88 | 106.0(12) | |
| C(15)–C(10)–C(11)–C(12) | –0.44 | –1.289 | 0(2) | C(10)–C(8)–C(9)–C(7) | 111.11 | 110.15 | –112.3(15) | |
| C(8)–C(10)–C(11)–C(12) | –179.67 | 0.68 | –179.6(13) | |||||
When the X-ray structure of the title compound is compared with its optimal counterparts, there are slight differences in compatibility between them, because experimental results are reported to solid phase while the theoretical calculations are related to the gaseous phase. In solid state, there is a crystalline field along with molecular interactions, leading to differences in the correlation parameters between calculated and experimental values. Globally, from Table 1, it can be observed that the computed geometrical parameters agreed very well with the single-crystal X-ray data. The average bond distance and bond angle in the three benzene rings for both experiment and calculation are in good agreement with literature values. The average C−F bond length in the cyclopropane ring is 1.36(2) Å and the C−C distances in this ring vary from 1.48(2) to 1.50(19) Å, which are in the expected range as found in earlier studies[30, 31]. The bond length C(16)=O(1) (1.22 Å) is ideal for tautomeric form Keto[32]. However, this distance is only shorter than C−OH (1.28 Å) of the enol form. The keto form is stabilized by intermolecular hydrogen bonds, which is not possible for the enol form. The mean values of the bond angles in the three benzene rings C(1)~C(6), C(10)~C(15) and C(17)~C(22) are 119.96°, 119.98° and 119.99°, respectively. For the cyclopropane ring, the mean bond angle is about 59.97°. The perspective view of the crystal packing in the unit cell is shown in Fig. 5.
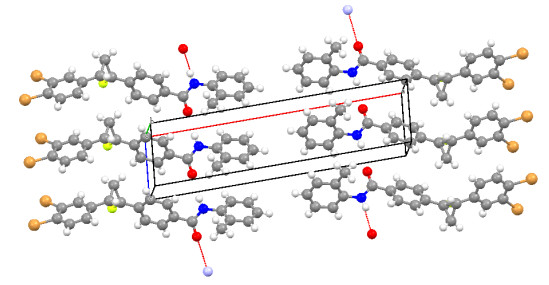
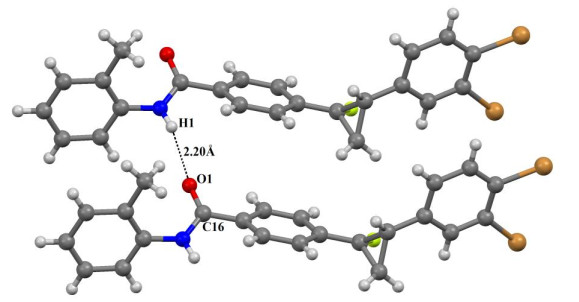
The compound involves intra- and intermolecular hydrogen bonding of C–H···O, N–H···O and C–H···F types in which C atoms (C(5) and C(12)) and N(1) act as donors and (O(1) and F(1)) atoms as acceptors. Fig. 6 shows N–H⋅⋅⋅O hydrogen bond in the crystal. This bond is formed due to the attraction between the oxygen atom (O(1)) of carbonyl (C(16)) and the hydrogen atom connected with nitrogen atom (N(1)). The details of H-bonds are shown in Table 2. These intermolecular interactions contribute to the stabilization of the crystal structure packing.
 DownLoad:
CSV
DownLoad:
CSV
| D–H···A | d(D–H) | d(H···A) | d(D···A) | ∠DHA |
| N(1)−H(1)···O(1)(i) | 0.86 | 2.20 | 2.939(11) | 144 |
| C(12)−H(12)···F(1)(ii) | 0.93 | 2.64 | 3.549(19) | 167 |
| C(5)−H(5)···O(1)(iii) | 0.93 | 2.59 | 3.406(19) | 147 |
| Symmetry codes: (i) x, y, z + 1, (ii) –x, y + 1/2, –z + 2, (iii) x, y, z–1 | ||||
For molecule containing N atoms, the number of normal modes of vibrations is 3N-6[33] in our case N=46. This number is 132. The scaled theoretical frequencies and IR intensity with the PED contributions at B3LYP/6-31G(d, p) are summarized in Table 3. The theoretical vibrational spectra IR and Raman with B3LYP/6-31G(d, p) and B3LYP/6-311++G(d, p) basis set are shown in Fig. 7.
DownLoad:
CSV
| Mode | Unscaled | Scaled | IR intensity | Vibration assignments (PED ≥ 10%) |
| 132 | 3605 | 3482 | 14.0979 | νNH (100%) |
| 131 | 3240 | 3121 | 0.1211 | νCH ring 1 (98%) |
| 130 | 3230 | 3112 | 1.847 | νCH ring 2 (97) |
| 129 | 3226 | 3111 | 4.988 | νCH ring 2 (98) |
| 128 | 3216 | 3098 | 2.26 | νCH ring 1 (93) |
| 127 | 3213 | 3094 | 0.0592 | νCH ring 2 (97) |
| 126 | 3201 | 3085 | 27.359 | νCH ring 3 (99) |
| 125 | 3193 | 3077 | 10.848 | νCH ring 2 (96) |
| 124 | 3187 | 3071 | 24.59 | νCH ring 3 (94) |
| 123 | 3186 | 3069 | 8.93 | νCH ring 1 (93) |
| 122 | 3176 | 3061 | 20.19 | νCH ring 2 (97) |
| 121 | 3173 | 3058 | 2.477 | νCH ring 3 (91) |
| 120 | 3168 | 3053 | 12.127 | νCH ring 3 (86) |
| 119 | 3156 | 3041 | 5.799 | νCH (99) |
| 118 | 3137 | 3024 | 5.004 | νCH2 (99) |
| 117 | 3122 | 3013 | 16.532 | νCH3 (98) |
| 116 | 3103 | 2993 | 13.553 | νCH3 (99) |
| 115 | 3039 | 2932 | 21.794 | νCH3 (97) |
| 114 | 1768 | 1700 | 165.79 | νOC (83) |
| 113 | 1666 | 1603 | 59.925 | νCC ring 2 (64) + δHCC ring 2 (16) |
| 112 | 1661 | 1598 | 18.41 | νCC ring 3 (51) + δHCC ring 3 (13) + δCCC ring 3 (12) |
| 111 | 1639 | 1576 | 5.804 | νCC ring 1 (55) + δHCC ring 1 (10) |
| 110 | 1638 | 1576 | 12.774 | νCC ring 3 (49) |
| 109 | 1616 | 1554 | 12.774 | νCC ring 2 (60) + δHCC ring 2 (13) + δCCC ring 2 (10) |
| 108 | 1600 | 1540 | 14.876 | νCC ring 1 (61) |
| 107 | 1556 | 1496 | 13.851 | δHCC ring 2 (37) |
| 106 | 1544 | 1484 | 16.376 | δHNC (11) + δCC R3 (30) + δCCC R3 (13) |
| 105 | 1528 | 1469 | 277.64 | δHNC (11) + δHCC ring 3 (16) + δCH3 (21) |
| 104 | 1511 | 1453 | 22.498 | δHCC ring 1 (31) |
| 103 | 1508 | 1450 | 43.15 | δHNC (12) + δCH3 (36) |
| 102 | 1493 | 1435 | 26.571 | δCH2 (56) |
| 101 | 1489 | 1430 | 27.778 | δCH3 (68) + τCCCH3 (12) |
| 100 | 1473 | 1417 | 113.77 | νCC ring 3 (14) + δHNC (12) + δHCC ring 3 (19) |
| 99 | 1446 | 1391 | 4.83 | νCC ring 2 (36) + δHCC ring 2 (15) + δCH2 (14) |
| 98 | 1443 | 1388 | 12.076 | νCC ring 2 (19) + δHCC Δ (17) + δHCC ring 2 (11) |
| 97 | 1428 | 1370 | 3.72 | δCH3 (90) |
| 96 | 1407 | 1353 | 9.88 | νCC ring 1 (13) + δHCC ring 1 (11) |
| 95 | 1357 | 1306 | 11 | νCC ring 2 (68) |
| 94 | 1350 | 1298 | 64 | νCC ring 3 (61) |
| 93 | 1341 | 1290 | 2.184 | δHCC ring 2 (65) |
| 92 | 1335 | 1284 | 0.268 | νCC ring 1 (44) + δHCC ring 2 (11) |
| 91 | 1315 | 1265 | 33.925 | νCC ring 1 (18) + νCC (19) |
| 90 | 1315 | 1264 | 61.155 | νCC ring 3 (11) + δHCC ring 3 (46) |
| 89 | 1290 | 1241 | 8.991 | νCC Δ (17) + δHCC Δ (12) + δHCC ring 1 (27) |
| 88 | 1285 | 1236 | 14.092 | νCC Δ (17) + δHCC ring 1 (28) |
| 87 | 1270 | 1222 | 119.66 | νCC ring 2 (37) + νNC (10) |
| 86 | 1264 | 1215 | 117.24 | νCC ring 3 (51) + δHNC (18) + δHCC ring R3 (10) |
| 85 | 1229 | 1182 | 6.642 | νCC (15) + δHCC Δ (13) + δCCC ring 1 (15) |
| 84 | 1222 | 1176 | 12.894 | νCC ring 3 (43) + δHCC ring 3 (19) |
| 83 | 1216 | 1169 | 17.488 | νCC ring 2 (16) + δHCC ring 2 (62) |
| 82 | 1190 | 1143 | 0.337 | νCC ring 3 (10) + δHCC ring 3 (79) |
| 81 | 1176 | 1130 | 2.746 | νCC ring 1 (11) + δHCC ring 1 (51) |
| 80 | 1151 | 1106 | 27.913 | δHCC ring 3 (12) + δHCC ring 2 (12) + δHCC ring 3 (13) |
| 79 | 1148 | 1104 | 6.561 | νCC ring 2 (12) + δHCC ring 2 (34) |
| 78 | 1143 | 1099 | 38.136 | δHCC ring 2 (14) + τHCCC Δ (12) + τHCCC Δ (16) |
| 77 | 1135 | 1091 | 14.121 | νCC ring 1 (46) + δHCC ring 1 (23) |
| 76 | 1118 | 1075 | 18.110 | νNC (26) + δCCC R3 (12) |
| 75 | 1092 | 1048 | 5.129 | τH CCC Δ (49) |
| 74 | 1077 | 1036 | 1.458 | νCC ring 3 (50) + δHCC ring 3 (12) |
| 73 | 1071 | 1027 | 3.629 | δCH3 (16) + τCCCH3 (52) |
| 72 | 1066 | 1022 | 38.567 | τHCCC Δ (53) + τHCCC Δ (12) |
| 71 | 1033 | 993 | 30.526 | δCCC ring 2 (66) |
| 70 | 1025 | 986 | 32.326 | νCC ring 1 (11) + δCCC ring 1 (73) |
| 69 | 1022 | 982 | 13.09 | νCC Δ (12) + δCCC Δ (24) + τHCCC Δ (25) |
| 68 | 1017 | 976 | 2.782 | δCH3 (11) + δCCC ring 3 (12) + τCCCH3 (48) |
| 67 | 1001 | 962 | 1.985 | τHCCC ring 2 (78) |
| 66 | 984 | 944 | 0.064 | τHCCC ring 3 (77) |
| 65 | 982 | 942 | 1.188 | τHCCC (61) + τCCCC (13) |
| 64 | 969 | 931 | 1.460 | δHCC (14) + τHCCC ring 2 (13) + τHCCC ring 1 (10) |
| 63 | 967 | 928 | 0.318 | τHCCC ring 2 (60) + τCCCC (13) |
| 62 | 947 | 909 | 2.779 | τHCCC ring 3 (68) τCCCN (10) |
| 61 | 919 | 883 | 3.003 | δOCN (12) + δCNC (10) τ+ HCCC ring 3 (15) |
| 60 | 917 | 882 | 21.721 | νCC Δ (21) + δHCC Δ (11) + τHCCC (31) |
| 59 | 914 | 880 | 2.327 | νCC ring 2 (14) + δHCC ring 2 (12) + τHCCC (39) |
| 58 | 888 | 853 | 2.751 | δHCC Δ (13) + τHCCC ring 2 (24) |
| 57 | 871 | 835 | 2.681 | τHCCC ring 3 (78) |
| 56 | 865 | 832 | 11.09 | νCC Δ (10) + δCCC ring 3 (19) + τHCCC ring 2 (10) |
| 55 | 860 | 826 | 5.697 | τHCCC ring 2 (32) + τHCCC ring 3 (29) |
| 54 | 850 | 815 | 9.570 | τHCCC ring 1 (79) |
| 53 | 840 | 807 | 32.519 | τHCCC ring 2 (42) |
| 52 | 814 | 782 | 20.861 | νCC Δ (10) + δCCC ring 1 (17) |
| 51 | 780 | 750 | 28.59 | νCC (15) + τHCCC ring 3 (22) |
| 50 | 769 | 739 | 21.182 | τCCCC ring 2 (13) + ωONCC (29) |
| 49 | 763 | 732 | 25.49 | τHCCC ring 3 (60) |
| 48 | 746 | 717 | 7.065 | τHCCC ring 1 (10) + ωCCCC (42) |
| 47 | 735 | 707 | 2.38 | δCCC ring 2 + (15) τCCCC ring 3 (–13) |
| 46 | 721 | 693 | 2.41 | ωCCCC ring 2 (11) + τCCCC ring 3 (34) |
| 45 | 707 | 680 | 16.799 | τCCCC ring 2 (30) + ωONCC (26) |
| 44 | 679 | 653 | 14.69 | δCCC ring 1 (50) |
| 43 | 676 | 649 | 13.30 | δCCC ring 1 (14) + ωFCCC (15) |
| 42 | 649 | 624 | 0.44 | νCC (10) + δCCC ring 2 (78) |
| 41 | 624 | 599 | 10.24 | δCCC ring 3 (44) |
| 40 | 614 | 590 | 4.469 | δCCC ring 3 (12) |
| 39 | 582 | 558 | 10.24 | ωCCCC ring 2 (22) + ωCCCC ring 1 (10) |
| 38 | 569 | 546 | 18.887 | τCCCC (–12) + τHNCC (–13) |
| 37 | 556 | 533 | 2.953 | δCCC (13) + τCCCC ring 3 (25) |
| 36 | 544 | 517 | 49.335 | νCC (12) + δCCC ring 3 (–26) + τHNCC (46) |
| 35 | 535 | 511 | 4.433 | τHNCC (59) + τCCCC ring 3 (25) |
| 34 | 493 | 473 | 6.076 | δFCC (23) |
| 33 | 473 | 455 | 5.459 | ωCCCC ring 2 (12) |
| 32 | 464 | 445 | 1.617 | τCCCC (49) |
| 31 | 458 | 440 | 8.098 | τCCCC ring 1 (27) + τCCCC ring 2 (19) |
| 30 | 445 | 428 | 1.711 | δCCC (12) + δCCN (18) |
| 29 | 441 | 423 | 3.507 | τCCCC ring 1 (17) |
| 28 | 420 | 404 | 1.652 | τHCCC ring 2 (11) + τHCCC ring 2 (13) + τCCCC ring 2 (64) |
| 27 | 406 | 389 | 0.321 | δCCC (13) |
| 26 | 403 | 387 | 4.697 | δCCBr (50) |
| 25 | 365 | 351 | 3.906 | δFCC (16) |
| 24 | 350 | 336 | 11.990 | νCC (39) |
| 23 | 330 | 316 | 0.5234 | δCCC (37) |
| 22 | 311 | 299 | 0.974 | νCC (11) + ωBrCCC (17) |
| 21 | 296 | 284 | 8.921 | δOCN (12) + δCCC (11) + τCCCN (25) |
| 20 | 277 | 265 | 3.050 | τCCCN (25) |
| 19 | 256 | 245 | 1.391 | ωCCCC (11) + ωCCCC (21) |
| 18 | 231 | 219 | 4.296 | δCCC (11) |
| 16 | 194 | 183 | 0.349 | νBrC (14) δCCC (19) |
| 15 | 186 | 168 | 1.42 | νBrC (14) + δCCC (19) |
| 14 | 172 | 163 | 1.027 | δNCC (14) + δCCC (10) + τHCCC (21) |
| 13 | 154 | 147 | 0.453 | τHCCC (10) + ωCCCC (13) + ωBrCCC (12) |
| 12 | 133 | 127 | 0.344 | ωCCCC (22) + ωBrCCC (12) |
| 11 | 119 | 112 | 1.968 | δBrCC (76) |
| 10 | 108 | 102 | 1.626 | ωCCCC (10) + τCNCC (40) |
| 9 | 104.6 | 100 | 1.851 | δCCC (13) + τCCCC (12) + τCNCC (20) |
| 8 | 83 | 79 | 0.237 | τCCCC (34) |
| 7 | 60.82 | 56 | 0.044 | δCCC (38) |
| 6 | 52.01 | 49 | 0.113 | τCNCC (53) |
| 5 | 48. | 46 | 0.118 | δCCC (11) + δCCC (17) + δCNC (12) + ωCCCC (15) |
| 4 | 34 | 30 | 0.144 | δCCC (12) + δCNC (33) |
| 3 | 20.93 | 20 | 0.090 | τCNCC (46) |
| 2 | 19.35 | 13 | 0.638 | δCNC (12) + τCCCC (55) |
| 1 | 14.54 | 11 | 0.0595 | τCNCC (30) + τCNCC (12) |
| ν: stretching, δ: bending, τ: twisting, γ: out of plane bending, s: symmetric, as: asymmetric | ||||
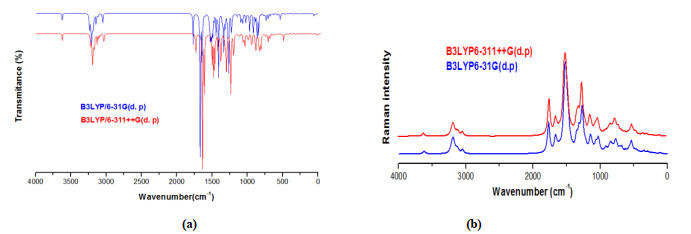
C−H aromatic stretching vibrations are varying 3100~3000 cm-1 that is the characteristic region for the ready identification of C−H stretching vibrations[34]. The peak of C−H stretching vibration is calculated in the region of 3041~3121 cm-1 by method B3LYP/6-31G (d, p) basis set. The maximum PED corresponding to this vibration contributes to 99%.
N−H stretching frequency band at 3605 cm-1 in FT-IR. The contribution of broad bands of PED is 100%.
The C−N stretching vibration is present in a composite region of the vibrational spectrum, that is to say, mixing of different bands is possible in this region[33]. The theoretical scaled frequency is calculated at 1075 and 1222 cm-1. The PED contribution is 26 and 12%, respectively.
C=O stretching is recorded at 1768 cm-1 in FT-IR, scaled frequencies at 1700 cm-1 with PED ≈ 83%. The carbonyl group of C=O stretching vibration (strong band) has characteristic band in the 1850~1550 cm-1 region[35].
We can see the aromatic benzene ring C−C bands at around 1650~1400 cm-1[35]. An aromatic ring replaces most of the ring modes. By referring the above notes, theoretical values of the title molecule were identified in the range of 1660~350 cm-1 via B3LYP/6-31G (d, p) method. The different frequencies in FI-IR were presented at 1603, 1598, 1576, 1554, 1540, 1306 and 1298 cm-1. The maximum contribution of PED of CC band is 68%.
Br−C stretching vibration gives generally strong band in the region of 650~485 cm-1[36-38]. In this compound, bands observed at 168 and 183 cm-1 for C−Br stretching as a mode (mode No. 16 and 15) have been assigned to C−Br in-plane and twisting vibration of CCCBr is observed at 147 cm-1 (mode No. 13). All these tasks are well aligned with calculated values and more justified with PED values.
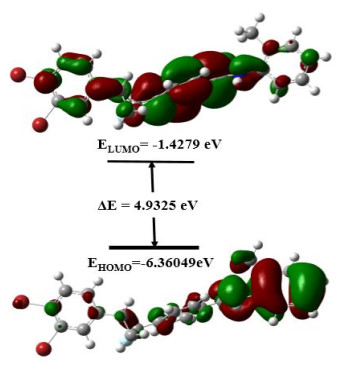
The frontier molecular orbitals are sketched in Fig. 8. The frontier orbital energy gap (ELUMO − EHOMO) of DBFB is found to be 4.9325 eV by B3LYP using 6-31G(d, p) basis set. This energy gap is a relatively high value, indicating significant stability of compounds that have biological activity. A lower HOMO-LUMO energy gap explains the fact that eventual charge transfer interaction is taking place within the molecule[39].
Both the global hardness and softness are concepts that have been used to explain chemical reactivity. According to Koopman theorem the chemical potential, hardness and softness can be written in terms of the frontier orbital energies (EHOMO and ELUMO). These two negative values define the ionization potential and electronic affinity, respectively. The calculated values of the global reactivity descriptors are listed in Table 4.
DownLoad:
CSV
| Parameters | Values (eV) |
| EHOMO | −6.226 |
| ELUMO | −1.395 |
| Gap (ΔE) | 4.832 |
| Electronegativity (χ) | 3.811 |
| Chemical potential (μ) | −3.811 |
| Chemical hardness (η) | 4.831 |
| Chemical softness (S) | 0.207 |
| Electrophilicity index (ω) | 1.503 |
| Additional electronic charge (ΔNmax) | 0.163 |
According to Parr an Pearson[40], electrophilicity index (ω) is as a global reactivity index similar to the chemical hardness and chemical potential. This reactivity index measures the stabilization in energy when the system acquires an additional electronic charge (ΔNmax) from the environment. The electrophilicity index (ω) is positive, definite quantity and the direction of the charge transfer is completely determined by the electronic chemical potential (μ) of the molecule because an electrophile is a chemical species capable of accepting electrons from the environment and its energy must decrease upon accepting electronic charge. Therefore, its electronic chemical potential must be negative. The structure stability of the title compound is confirmed by the negative value of the chemical potential (μ = −3.811 eV). The calculated value of chemical hardness (η = 4.831 eV) indicates that the charge transfer occurs within the molecule.
Computed Mulliken atomic charges play an important role in the application of quantum chemical computation to molecular polarizability, electronic structure and other properties of molecular systems[41]. Our calculated Mulliken charge values using HF and B3LYP with 6-31G (d, p) basis sets are gathered in Table 5. The results of Table 5 reveal the effect of DFT method and HF in the value of Mulliken charge distribution but the same behavior is observed. In fact, the Mulliken atomic charge analysis of DBFB shows that the fluorine atom has a maximum negative charge value of –0.3015 at DFT/6-31G(d, p). With the same level of theory, the maximum positive atomic charge of 0.795130 is obtained for C(16) which was imposed by O(1). However, atoms C(3), C(5), C(6), C(7), C(9), C(11), C(12) and C(14) possess small negative charges, whereas C(1) and C(2) exhibit a positive charge due to negative charge of Br(1) and Br(2). Moreover, all the hydrogen atoms have a net positive charge (for all the considered theoretical approaches).
DownLoad:
CSV
| Atoms | Mulliken atomic charges | Atoms | Mulliken atomic charges | |||
| DFT | HF | DFT | HF | |||
| Br(1) | –0.094459 | –0.075057 | C(16) | 0.553734 | 0.795130 | |
| Br(2) | –0.093303 | –0.073689 | C(21) | –0.098879 | –0.159134 | |
| N(1) | –0.639660 | –0.808507 | C(22) | –0.10338 | –0.142897 | |
| F(1) | –0.301539 | –0.400608 | C(23) | –0.385374 | –0.346166 | |
| O(1) | –0.503772 | –0.594833 | C(19) | –0.139060 | –0.166692 | |
| C(1) | 0.039286 | –0.027181 | C(20) | –0.074642 | –0.140331 | |
| C(2) | 0.036221 | –0.021984 | H(4) | 0.261137 | 0.309928 | |
| C(3) | –0.139351 | –0.122195 | H(3) | 0.116343 | 0.189077 | |
| C(4) | 0.196518 | 0.059095 | H(5) | 0.094871 | 0.164301 | |
| C(5) | –0.131284 | –0.157487 | H(6) | 0.116593 | 0.185202 | |
| C(6) | –0.088779 | –0.111020 | H(7) | 0.111783 | 0.157378 | |
| C(7) | –0.203012 | –0.214280 | H(9B) | 0.141130 | 0.165981 | |
| C(8) | 0.297273 | 0.445493 | H(9A) | 0.126005 | 0.149713 | |
| C(9) | –0.248240 | –0.283870 | H(15) | 0.112567 | 0.189030 | |
| C(10) | 0.116477 | –0.032461 | H(14) | 0.124193 | 0.201208 | |
| C(15) | –0.128951 | –0.154107 | H(11) | 0.086824 | 0.15351 | |
| C(14) | –0.100554 | –0.111773 | H(20) | 0.084919 | 0.151090 | |
| C(13) | 0.038415 | –0.151646 | H(21) | 0.086260 | 0.151493 | |
| C(12) | –0.132461 | –0.142267 | H(22) | 0.079983 | 0.151043 | |
| C(11) | –0.146731 | –0.176673 | H(23) | 0.109964 | 0.12170 | |
| H(19) | 0.082299 | 0.148166 | H(23B) | 0.162564 | 0.171392 | |
| C(17) | 0.232932 | 0.247133 | H(23A) | 0.100062 | 0.115260 | |
| C(18) | 0.151961 | 0.026484 | H(12) | 0.093125 | 0.166038 | |
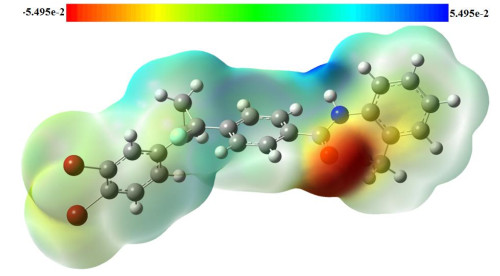
The chemical reactivity of the molecules can be predicted by using the MEP details[42]. Both experimental X-ray diffraction and theoretical methods are used to obtain MEP values[43, 44]. Total B3LYP/6-31G(d, p) electron density surface mapped with MEP of the DBFB molecule is shown in Fig. 9. The MEP displays molecular shape, size and electrostatic potential values. The extreme limits of the total electron density lay in the range of –5.495~5.495 e–2. The electron density varies significantly around the DBFB of electron withdrawing and donating groups. The color scheme for the MEP surface is blue-electron deficient or partially positive charge; red-electron rich or partially negative charge; light blue-slightly electron deficient region; yellow-slightly electron rich region, respectively. The title compound has several possible sites for electrophilic at O, N, F and Br atoms. The MEP map shows that the negative potential sites are on electronegative atoms and the positive potential sites are around the hydrogen atoms. These active sites are found to be clear evidence of biological activity in DBFB and give information about the region from which the compound can have intermolecular interactions.
In the present study, X-ray structure of a pyrethroid derivative has been determined. This compound belongs to the monoclinic system with P21 space group. Intramolecular C–H⋅⋅⋅O, N–H⋅⋅⋅O and C–H⋅⋅⋅F hydrogen bonds are present in the crystal structure. These hydrogen bonds contribute to the stability of the crystal packing. The stability of the compound has been confirmed by conformational analysis, indicating that our molecule exhibits trans isomer. Furthermore, theoretical calculations were carried out on the most stable conformer of the title molecule. The bond lengths and angles computed at the B3LYP/6-31G (d, p) level of theory were compared with the experimental ones, which are consistent with each other. Vibrational wavenumbers of the molecule have been calculated and assigned using the same level of theory with two different bases sets. Results show a good correlation with the structurally similar compounds. The HOMO-LUMO energy gap explains the charge transfer interactions and the eventual electronic transition taking place within the molecule. The calculated HOMO and LUMO energies were used to estimate the ionization potential, electron affinity, electronegativity, electrophilicity index, hardness and chemical potential. Moreover, Mulliken charges and molecular electrostatic potential were calculated to know about the potential and charge distribution within the molecule.
Cruz-Cabeza, A. J.; Allen F. H. Conformation and geometry of cyclopropane rings having π-acceptor substituents: a theoretical and database study. Acta Cryst. B 2011, 67, 94–102. doi: 10.1107/S0108768110049517
Hamzaoui, F.; Chouaih, A.; Lagant, P.; Belarbi, O.; Vergoten, G. A comparative X-ray diffraction study and ab initio calculation on RU60358, a new pyrethroid. Int. J. Mol. Sci. 2006, 7, 255–265. doi: 10.3390/i7080255
Baert, F.; Guelzim, A. X-ray structure of the pyrethroid insecticide {1R-[1α(S*), 2α]}-2-(2, 2-dichlorovinyl)-3, 3-dimethyl cyclopropane carboxylic acid cyano(3-phenoxyphenyl) methyl ester (RU 24501). Acta Cryst. C 1991, 47, 606–608.
Hamzaoui, F.; Lamiot, J.; Baert, F. X-ray structure of a new pyrethroid, RU 52259. Acta Cryst. C 1993, 49, 818–820.
Brooks, I. C.; Haus, J.; Blumenthal, R. R.; Davis Jr, B. S. SBP-1382, a new synthetic pyrethroid. Soap Chem. Spec. 1969, 45, 62–64.
Hill, A. S.; McAdam, D. P.; Edward, S. L.; Skerritt, J. H. Quantitation of bioresmethrin, a synthetic pyrethroid grain protectant, by enzyme immunoassay. J. Agric. Food Chem. 1993, 41, 2011–2018. doi: 10.1021/jf00035a038
Qu, J. P.; Deng, C.; Zhou, J.; Sun, X. L.; Tang, Y. Switchable reactions of cyclopropanes with enol silyl ethers. Controllable synthesis of cyclopentanes and 1, 6-dicarbonyl compounds. J. Org. Chem. 2009, 74, 7684–7689. doi: 10.1021/jo901340v
Hu, B.; Xing, S.; Ren, J.; Wang, Z. Total synthesis of (±)-bruguierol A via an intramolecular [3+2] cycloaddition of cyclopropane 1, 1-diester. Tetrahedron 2010, 66, 5671–5674. doi: 10.1016/j.tet.2010.05.057
Bhanot, S. K.; Singh, M.; Chatterjee, N. R. The chemical and biological aspects of fluoroquinolones reality and dreams. Curr. Pharm. Des. 2001, 7, 331–335.
Boger, D. L.; Hughes, T. V.; Hedrick, M. P. Synthesis, chemical properties, and biological evaluation of CC-1065 and duocarmycin analogues incorporating the 5-methoxycarbonyl-1, 2, 9, 9a-tetrahydrocyclopropa [c]benz[e] indol-4-one alkylation subunit. J. Org. Chem. 2001, 66, 2207–2216. doi: 10.1021/jo001772g
Ellis, D.; Kuhen, K. L.; Anaclerio, B.; Wu, B.; Wolff, K.; Yin, H.; Bursulaya, B.; Caldwell, J.; Karanewsky, D.; He, Y. Design, synthesis, and biological evaluations of novel quinolones as HIV-1 non-nucleoside reverse transcriptase inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 4246–4251. doi: 10.1016/j.bmcl.2006.05.073
Hamzaoui, F.; Baert, F. A new pyrethroid insecticide, RU41414. Acta Cryst. C 1996, 52, 689–690. doi: 10.1107/S0108270195013102
Elliott, M. The relationship between the structure and the activity of pyrethroids. Bull. Wld Hlth Org. 1970, 44, 315–324.
Elliott, M.; Farnham, A. W.; Janes, N. F.; Needham, P. H.; Pulman, D. A. Insecticidal activity of the pyrethrins and related compounds. Pestic. Sci. 1975, 6, 537–542. doi: 10.1002/ps.2780060514
Ali, R.; Fatemeh, Z. N.; Younes, H.; Sang Woo, J.; Masoome, S.; Katarzyna, Ś.; Tadeusz, L.; Farideh, G. Synthesis, crystal structure and theoretical calculations of N-benzyl-1-(5-(3-chlorophenyl)-1, 3, 4-oxadiazol-2-yl)cyclopentanamine. Chin. J. Struct. Chem. 2018, 37, 679–692.
Zhai, Z. W.; Shi, Y. X.; Yang, M. Y.; Sun, Z. H.; Weng, J. Q.; Tan, C. X.; Liu, X. H.; Li, B. J.; Zhang, Y. G. Synthesis, crystal structure, DFT studies and antifungal activity of 5-(4-cyclopropyl-5-((3-fluorobenzyl)sulfonyl)-4H-1, 2, 4-triazol-3-yl)-4-methyl-1, 2, 3-thiadiazole. Chin. J. Struct. Chem. 2016, 35, 25–33.
Hooriye, Y.; Ali Reza, K.; Ali, R. Synthesis and chemical shifts calculation of α-acyloxycarboxamides derived from indane-1, 2, 3-trione by DFT and HF methods. Chin. J. Struct. Chem. 2012, 31, 1346–1356.
Sheldrick, G. M. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. doi: 10.1107/S2053229614024218
Becke, A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. doi: 10.1063/1.464913
Lee, C.; Yang, W.; Parr, R. G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. doi: 10.1103/PhysRevB.37.785
Cohen, H. D.; Roothaan, C. C. J. Electric dipole polarizability of atoms by the Hartree-Fock method. I. Theory for closed-shell systems. J. Chem. Phys. 1965, 43, 34–39. doi: 10.1063/1.1701512
Jamroz, M. H. Vibrational energy distribution analysis (VEDA): scopes and limitations. Spectrochim. Acta A 2004, 114, 220–230.
Rauhut, G.; Pulay, P. Transferable scaling factors for density functional derived vibrational force fields. J. Phys. Chem. 1995, 99, 3093–3100. doi: 10.1021/j100010a019
Gómez Marigliano, A. C.; Varetti, E. L. Self-association of formamide in carbon tetrachloride solutions: an experimental and quantum chemistry vibrational and thermodynamic study. J. Phys. Chem. A 2002, 106, 1100–1106. doi: 10.1021/jp011060+
Dimitrova, Y.; Tsenov, J. A. Theoretical study of the structures and vibrational spectra of the hydrogen-bonded systems of 4-cyanophenol with N-bases. Spectrochim. Acta Part A 2007, 68, 454–459. doi: 10.1016/j.saa.2006.11.050
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenberg, J. L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, J. A. Jr.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, J. M.; Klene, M.; Knox, J. E.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Dapprich, S.; Daniels, A. D.; Farkas, Ö.; Foresman, J. B.; Ortiz, J. V.; Cioslowski, J.; Fox, D. J. Gaussian 09, Revision A. 02. Gaussian, Inc., Pittsburgh PA 2009.
Allegretti, P. E.; Milazzo, C. B.; Castro, E. A.; Furlong J. J. P. Mass spectrometry as a valuable tool for the study of tautomerism of amides and thioamides. J. Mol. Struct. (THEOCHEM. ) 2002, 589–590, 161–170.
Yahiaoui, S.; Chouaih, A.; Hamzaoui, F. X-ray and DFT crystal structure determination and conformational analysis of a pyrethroid compound. Chin. J. Struct. Chem. 2013, 32, 1544–1552.
Singh, H.; Singh, S.; Srivastava, A.; Tandon, P.; Bharti, P.; Kumar, S.; Maurya, R. Conformational analysis and vibrational study of daidzein by using FT-IR and FT-Raman spectroscopies and DFT calculations. Spectrochem. Acta Part A: Mol. Biomol. Spectrosc. 2014, 120, 405–415. doi: 10.1016/j.saa.2013.10.045
Tessier, J.; Teche, A.; Demoute, J. P. Proceedings of the 5th IUPAC International Congress of Pesticide Chemistry. J. Miyamoto, P. C. Kearney, Ed., Pergamon Press: Oxford, New York 1983, 197–202.
Tessier, J.; Teche, A.; Demoute, J. P. Pesticide Chemistry: Human Welfare and the Environment. J. Miyamoto, P. C. Kearney Ed., Pergamon Press: Oxford, New York 1983, 1, 95–100.
Sharma, A.; Jad, Y. E.; Ghabbour, H. A.; De la Torre, B. G.; Kruger, H. G.; Albericio, F.; El-Faham, A. Synthesis, crystal structure and DFT studies of 1, 3-dimethyl-5-propionylpyrimidine-2, 4, 6(1H, 3H, 5H)-trione. Crystals 2017, 7, 31–40. doi: 10.3390/cryst7010031
Silverstein, M.; Bassler, G. C.; Morril, C. Spectroscopic Identification of Organic Compounds. John Wiley: New York 1981.
Subashchandrabose, S.; Saleem, H.; Erdogdu, Y.; Dereli, O.; Thanikachalam, V.; Jayabharathi, J. Structural, vibrational and hyperpolarizability calculation of (E)-2-(2-hydroxybenzylideneamino)-3-methylbutanoic acid. Spectrochem. Acta Part A: Mol. Biomol. Spectrosc. 2012, 86, 231–241. doi: 10.1016/j.saa.2011.10.029
Varsanyi, G. Assignment for Vibrational Spectra of Seven Hundred Benzene Derivatives. Academic Kiaclo: Budapest 1973.
Sortur, V.; Yenagi, J.; Tonannavar, J.; Jadhav, V. B.; Kulkarni, M. V. Vibrational assignments for 7-methyl-4-bromomethylcoumarin, as aided by RHF and B3LYP/6-31G* calculations. Spectrochem. Acta Part A: Mol. Biomol. Spectrosc. 2008, 71, 688–694. doi: 10.1016/j.saa.2008.01.016
Risgin, J. H. Fluorocarbons and related compounds, Vol. II. Academic press: New York 1954, 449–452.
Shakila, G.; Saleem H.; Sundaraganesan, N. FT-IR, FT-Raman, NMR and U-V spectral investigation: computation of vibrational frequency, chemical shifts and electronic structure calculations of 1-bromo-4-nitrobenzene. World Scientific News 2017, 61, 150–185.
Seminario, J. M. Recent Developments and Applications of Modern Density Functional Theory, Vol. 4. Elsevier 1996, pp. 800–806.
Parr, R. G.; Pearson, R. G. Absolute hardness: comparison parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. doi: 10.1021/ja00364a005
Mulliken, R. S. Electronic population analysis on LCAO-MO molecular wave functions. J. Chem. Phys. 1955, 23 1833–1840. doi: 10.1063/1.1740588
Drissi, M.; Benhalima, N.; Megrouss, Y.; Rahmani, R.; Chouaih, A.; Hamzaoui, F. Theoretical and experimental electrostatic potential around the m-nitrophenol molecule. Molecules 2015, 20, 4042–4054. doi: 10.3390/molecules20034042
Megrouss, Y.; Benhalima, N.; Bahoussi R.; Boukabcha, N.; Chouaih, A.; Hamzaoui, F. Determination of electrostatic parameters of a coumarin derivative compound C17H13NO3 by X-ray and density functional theory. Chin. Phys. B 2015, 24, 106103–7. doi: 10.1088/1674-1056/24/10/106103
Boubegra, N.; Chouaih, A.; Drissi, M.; Hamzaoui, F. Structural and electron charge density studies of a nonlinear optical compound 4, 4 di-methyl amino cyano biphenyl. Chin. Phys. B 2014, 23, 016103–6. doi: 10.1088/1674-1056/23/1/016103

Figure 4 Geometry of DBFB (a) X-ray structure with atomic numbering scheme and (b) optimized molecular structure

Figure 9 Total electron density surface mapped with molecular electrostatic potential of DBFB
Table 1. Geometrical Parameters of DBFB
| Bond | Dist. (Å) | Bond | Dist. (Å) | |||||
| HF | B3LYP | X-ray | HF | B3LYP | X-ray | |||
| Br(1)–C(1) | 1.897 | 1.900 | 1.854(12) | C(17)–C(22) | 1.386 | 1.400 | 1.40(3) | |
| Br(2)–C(2) | 1.898 | 1.903 | 1.858(15) | C(17)–C(18) | 1.395 | 1.409 | 1.36(2) | |
| N(1)–C(16) | 1.354 | 1.383 | 1.315(13) | F(1)–C(8) | 1.360 | 1.381 | 1.36(2) | |
| N(1)–C(17) | 1.427 | 1.426 | 1.410(13) | C(13)–C(14) | 1.390 | 1.400 | 1.336(17) | |
| N(1)–H(1) | 0.994 | 1.010 | 0.8600 | C(7)–C(9) | 1.508 | 1.520 | 1.48(2) | |
| C(16)–O(1) | 1.202 | 1.224 | 1.224(13) | C(7)–C(8) | 1.508 | 1.531 | 1.501(19) | |
| C(16)–C(13) | 1.501 | 1.503 | 1.467(13) | C(14)–C(15) | 1.382 | 1.391 | 1.373(16) | |
| C(4)–C(3) | 1.387 | 1.401 | 1.371(19) | C(1)–C(6) | 1.385 | 1.395 | 1.30(3) | |
| C(4)–C(5) | 1.390 | 1.402 | 1.38(2) | C(1)–C(2) | 1.385 | 1.398 | 1.41(2) | |
| C(4)–C(7) | 1.496 | 1.490 | 1.483(13) | C(2)–C(3) | 1.385 | 1.394 | 1.365(17) | |
| C(12)–C(11) | 1.389 | 1.392 | 1.382(16) | C(10)–C(15) | 1.391 | 1.402 | 1.382(19) | |
| C(12)–C(13) | 1.387 | 1.402 | 1.41(2) | C(10)–C(11) | 1.390 | 1.402 | 1.37(2) | |
| C(19)–C(20) | 1.386 | 1.394 | 1.40(3) | C(10)–C(8) | 1.498 | 1.490 | 1.468(15) | |
| C(19)–C(18) | 1.390 | 1.401 | 1.414(18) | C(8)–C(9) | 1.485 | 1.496 | 1.48(2) | |
| C(21)–C(22) | 1.385 | 1.393 | 1.37(2) | C(18)–C(23) | 1.509 | 1.506 | 1.48(3) | |
| C(21)–C(20) | 1.389 | 1.394 | 1.37(3) | C(6)–C(5) | 1.380 | 1.391 | 1.39(2) | |
| Bond angle (°) | HF | B3LYP | X-ray | Bond angle (°) | HF | B3LYP | X-ray | |
| C(16)–N(1)–C(17) | 123.32 | 124.67 | 125.2(9) | C(3)–C(2)–C(1) | 120.26 | 120.34 | 118.0(14) | |
| C(17)–N(1)–H(1) | 116.41 | 115.34 | 117.4(10) | C(3)–C(2)–Br(2) | 117.25 | 117.49 | 118.5(12) | |
| O(1)–C(16)–N(1) | 122.83 | 122.85 | 122.5(9) | C(1)–C(2)–Br(2) | 122.42 | 122.15 | 123.5(11) | |
| O(1)–C(16)–C(13) | 121.05 | 121.53 | 121.2(9) | C(4)–C(3)–C(2) | 118.21 | 120.90 | 123.6(14) | |
| N(1)–C(16)–C(13) | 116.10 | 115.60 | 116.3(9) | C(15)–C(10)–C(11) | 118.21 | 118.71 | 118.9(11) | |
| C(3)–C(4)–C(5) | 120.35 | 118.13 | 116.6(12) | C(15)–C(10)–C(8) | 119.99 | 120.00 | 120.8(13) | |
| C(3)–C(4)–C(7) | 123.30 | 123.12 | 126.0(12) | C(11)–C(10)–C(8) | 121.18 | 121.11 | 120.3(12) | |
| C(5)–C(4)–C(7) | 118.48 | 118.73 | 117.4(13) | C(14)–C(15)–C(10) | 120.42 | 120.51 | 119.7(13) | |
| C(11)–C(12)–C(13) | 120.54 | 120.63 | 119.5(13) | C(14)–C(15)–H(15) | 119.51 | 120.18 | 120.1 | |
| C(20)–C(19)–C(18) | 121.65 | 121.99 | 121.2(18) | C(10)–C(11)–C(12) | 120.62 | 120.65 | 120.6(13) | |
| C(22)–C(21)–C(20) | 119.34 | 119.44 | 116.9(18) | F(1)–C(8)–C(10) | 111.73 | 112.09 | 112.7(12) | |
| C(22)–C(17)–N(1) | 118.58 | 117.94 | 118.8(15) | F(1)–C(8)–C(9) | 113.89 | 114.28 | 112.3(13) | |
| C(22)–C(17)–C(18) | 121.73 | 120.81 | 120.2(14) | C(10)–C(8)–C(9) | 124.81 | 124.60 | 124.4(13) | |
| N(1)–C(17)–C(18) | 120.60 | 121.63 | 120.9(14) | F(1)–C(8)–C(7) | 114.44 | 114.51 | 114.3(9) | |
| C(14)–C(13)–C(12) | 118.78 | 118.60 | 118.5(10) | C(10)–C(8)–C(7) | 122.57 | 121.72 | 123.8(12) | |
| C(14)–C(13)–C(16) | 118.01 | 117.62 | 121.0(11) | C(9)–C(8)–C(7) | 58.97 | 58.66 | 59.5(12) | |
| C(12)–C(13)–C(16) | 123.08 | 123.68 | 120.4(11) | C(19)–C(20)–C(21) | 119.86 | 119.71 | 121.1(14) | |
| C(9)–C(7)–C(4) | 124.54 | 123.33 | 120.1(12) | C(17)–C(18)–C(19) | 117.75 | 117.52 | 117.0(17) | |
| C(9)–C(7)–C(8) | 58.97 | 58.66 | 59.6(12) | C(17)–C(18)–C(23) | 121.73 | 122.19 | 124.3(13) | |
| C(4)–C(7)–C(8) | 121.98 | 122.97 | 121.8(11) | C(19)–C(18)–C(23) | 120.52 | 120.40 | 118.7(16) | |
| C(13)–C(14)–C(15) | 120.78 | 120.89 | 122.3(12) | C(1)–C(6)–C(5) | 120.97 | 120.30 | 123.8(17) | |
| C(6)–C(1)–C(2) | 119.13 | 119.15 | 118.3(13) | C(21)–C(22)–C(17) | 120.59 | 120.75 | 123.4(19) | |
| C(6)–C(1)–Br(1) | 117.87 | 118.75 | 122.5(13) | C(4)–C(5)–C(6) | 121.06 | 121.15 | 119.3(17) | |
| C(2)–C(1)–Br(1) | 122.99 | 122.08 | 119.2(14) | C(7)–C(9)–C(8) | 60.48 | 61.11 | 60.8(8) | |
| Dihedral angle (°) | HF | B3LYP | X-ray | Dihedral angle (°) | HF | B3LYP | X-ray | |
| C(17)–N(1)–C(16)–C(13) | 176.80 | 175.64 | –176.6(15) | C(15)–C(10)–C(8)–F(1) | 8.47 | 7.94 | –13.1(17) | |
| C(17)–N(1)–C(16)–O(1) | –3.37 | –4.14 | 2(2) | C(11)–C(10)–C(8)–F(1) | –172.55 | –173.02 | 166.1(12) | |
| C(16)–N(1)–C(17)–C(22) | –111.92 | –121.09 | 118.5(17) | C(15)–C(10)–C(8)–C(9) | 152.26 | 152.54 | −154.7(18) | |
| C(16)–N(1)–C(17)–C(18) | 70.73 | 62.16 | –66(2) | C(11)–C(10)–C(8)–C(9) | –28.68 | –28.22 | 25(2) | |
| C(11)–C(12)–C(13)–C(14) | –1.08 | 1.06 | –6(2) | C(15)–C(10)–C(8)–C(7) | –133.32 | –133.87 | 131.6(15) | |
| C(11)–C(12)–C(13)–C(16) | –179.64 | 179.06 | 178.6(12) | C(11)–C(10)–C(8)–C(7) | 45.90 | 45.17 | –49.1(16) | |
| O(1)–C(16)–C(13)–C(14) | 24.93 | 22.47 | –34.4(18) | C(9)–C(7)–C(8)–F(1) | –105.64 | 106.26 | –102.5(14) | |
| N(1)–C(16)–C(13)–C(14) | –154.85 | –157.60 | 144.3(13) | C(4)–C(7)–C(8)–F(1) | –7.59 | –6.60 | 6.2(16) | |
| O(1)–C(16)–C(13)–C(12) | –153.17 | –156.24 | 140.9(13) | C(9)–C(7)–C(8)–C(10) | 111.11 | –113.93 | 113.2(17) | |
| N(1)–C(16)–C(13)–C(12) | 26.22 | 23.70 | –40.4(17) | C(4)–C(7)–C(8)–C(10) | 131.48 | 134.20 | –138.0(13) | |
| C(3)–C(4)–C(7)–C(9) | –13.27 | –30.20 | 16(2) | C(4)–C(7)–C(8)–C(9) | –112.49 | –111.87 | 108.7(15) | |
| C(5)–C(4)–C(7)–C(9) | 166.77 | 149.42 | –161.5(17) | N(1)–C(17)–C(18)–C(19) | 178.88 | 178.30 | 179.2(12) | |
| C(3)–C(4)–C(7)–C(8) | 58.80 | 41.50 | –55.2(16) | C(22)–C(17)–C(18)–C(23) | –179.01 | –178.38 | 174.3(19) | |
| C(5)–C(4)–C(7)–C(8) | –121.15 | –138.83 | 127.7(15) | C(20)–C(19)–C(18)–C(23) | 178.85 | 177.54 | –176.3(17) | |
| C(12)–C(13)–C(14)–C(15) | –1.08 | –1.42 | 7(2) | C(2)–C(1)–C(6)–C(5) | 0.09 | –0.11 | 8(3) | |
| C(16)–C(13)–C(14)–C(15) | –179.64 | 179.79 | –177.2(13) | Br(1)–C(1)–C(6)–C(5) | 179.95 | 179.88 | –174.7(16) | |
| Br(1)–C(1)–C(2)–C(3) | –179.82 | –179.85 | 175.2(10) | C(20)–C(21)–C(22)–C(17) | –0.58 | –0.57 | –3(3) | |
| C(6)–C(1)–C(2)–Br(2) | 179.80 | –179.87 | 174.0(15) | N(1)–C(17)–C(22)–C(21) | –178.20 | –177.61 | –179.0(18) | |
| Br(1)–C(1)–C(2)–Br(2) | 0.08 | 0.16 | –3.5(17) | C(18)–C(17)–C(22)–C(21) | –0.15 | –0.77 | 5(3) | |
| C(7)–C(4)–C(3)–C(2) | –179.77 | 179.27 | –176.2(12) | C(3)–C(4)–C(5)–C(6) | –0.06 | 0.39 | –1(2) | |
| C(1)–C(2)–C(3)–C(4) | –0.17 | 0.12 | 3(2) | C(7)–C(4)–C(5)–C(6) | 179.90 | –179.25 | 176.6(16) | |
| Br(2)–C(2)–C(3)–C(4) | 179.97 | –179.80 | –178.2(11) | C(4)–C(7)–C(9)–C(8) | 109.66 | –111.86 | –111.4(13) | |
| C(8)–C(10)–C(15)–C(14) | 179.08 | 0.984 | –179.1(13) | F(1)–C(8)–C(9)–C(7) | –105.64 | –105.88 | 106.0(12) | |
| C(15)–C(10)–C(11)–C(12) | –0.44 | –1.289 | 0(2) | C(10)–C(8)–C(9)–C(7) | 111.11 | 110.15 | –112.3(15) | |
| C(8)–C(10)–C(11)–C(12) | –179.67 | 0.68 | –179.6(13) | |||||
 下载: 导出CSV
下载: 导出CSV
Table 2. Hydrogen Bond Lengths (Å) and Bond Angles (°)
| D–H···A | d(D–H) | d(H···A) | d(D···A) | ∠DHA |
| N(1)−H(1)···O(1)(i) | 0.86 | 2.20 | 2.939(11) | 144 |
| C(12)−H(12)···F(1)(ii) | 0.93 | 2.64 | 3.549(19) | 167 |
| C(5)−H(5)···O(1)(iii) | 0.93 | 2.59 | 3.406(19) | 147 |
| Symmetry codes: (i) x, y, z + 1, (ii) –x, y + 1/2, –z + 2, (iii) x, y, z–1 | ||||
下载: 导出CSV
Table 3. Theoretical Vibrational Wavenumbers (cm–1) for DBFB
| Mode | Unscaled | Scaled | IR intensity | Vibration assignments (PED ≥ 10%) |
| 132 | 3605 | 3482 | 14.0979 | νNH (100%) |
| 131 | 3240 | 3121 | 0.1211 | νCH ring 1 (98%) |
| 130 | 3230 | 3112 | 1.847 | νCH ring 2 (97) |
| 129 | 3226 | 3111 | 4.988 | νCH ring 2 (98) |
| 128 | 3216 | 3098 | 2.26 | νCH ring 1 (93) |
| 127 | 3213 | 3094 | 0.0592 | νCH ring 2 (97) |
| 126 | 3201 | 3085 | 27.359 | νCH ring 3 (99) |
| 125 | 3193 | 3077 | 10.848 | νCH ring 2 (96) |
| 124 | 3187 | 3071 | 24.59 | νCH ring 3 (94) |
| 123 | 3186 | 3069 | 8.93 | νCH ring 1 (93) |
| 122 | 3176 | 3061 | 20.19 | νCH ring 2 (97) |
| 121 | 3173 | 3058 | 2.477 | νCH ring 3 (91) |
| 120 | 3168 | 3053 | 12.127 | νCH ring 3 (86) |
| 119 | 3156 | 3041 | 5.799 | νCH (99) |
| 118 | 3137 | 3024 | 5.004 | νCH2 (99) |
| 117 | 3122 | 3013 | 16.532 | νCH3 (98) |
| 116 | 3103 | 2993 | 13.553 | νCH3 (99) |
| 115 | 3039 | 2932 | 21.794 | νCH3 (97) |
| 114 | 1768 | 1700 | 165.79 | νOC (83) |
| 113 | 1666 | 1603 | 59.925 | νCC ring 2 (64) + δHCC ring 2 (16) |
| 112 | 1661 | 1598 | 18.41 | νCC ring 3 (51) + δHCC ring 3 (13) + δCCC ring 3 (12) |
| 111 | 1639 | 1576 | 5.804 | νCC ring 1 (55) + δHCC ring 1 (10) |
| 110 | 1638 | 1576 | 12.774 | νCC ring 3 (49) |
| 109 | 1616 | 1554 | 12.774 | νCC ring 2 (60) + δHCC ring 2 (13) + δCCC ring 2 (10) |
| 108 | 1600 | 1540 | 14.876 | νCC ring 1 (61) |
| 107 | 1556 | 1496 | 13.851 | δHCC ring 2 (37) |
| 106 | 1544 | 1484 | 16.376 | δHNC (11) + δCC R3 (30) + δCCC R3 (13) |
| 105 | 1528 | 1469 | 277.64 | δHNC (11) + δHCC ring 3 (16) + δCH3 (21) |
| 104 | 1511 | 1453 | 22.498 | δHCC ring 1 (31) |
| 103 | 1508 | 1450 | 43.15 | δHNC (12) + δCH3 (36) |
| 102 | 1493 | 1435 | 26.571 | δCH2 (56) |
| 101 | 1489 | 1430 | 27.778 | δCH3 (68) + τCCCH3 (12) |
| 100 | 1473 | 1417 | 113.77 | νCC ring 3 (14) + δHNC (12) + δHCC ring 3 (19) |
| 99 | 1446 | 1391 | 4.83 | νCC ring 2 (36) + δHCC ring 2 (15) + δCH2 (14) |
| 98 | 1443 | 1388 | 12.076 | νCC ring 2 (19) + δHCC Δ (17) + δHCC ring 2 (11) |
| 97 | 1428 | 1370 | 3.72 | δCH3 (90) |
| 96 | 1407 | 1353 | 9.88 | νCC ring 1 (13) + δHCC ring 1 (11) |
| 95 | 1357 | 1306 | 11 | νCC ring 2 (68) |
| 94 | 1350 | 1298 | 64 | νCC ring 3 (61) |
| 93 | 1341 | 1290 | 2.184 | δHCC ring 2 (65) |
| 92 | 1335 | 1284 | 0.268 | νCC ring 1 (44) + δHCC ring 2 (11) |
| 91 | 1315 | 1265 | 33.925 | νCC ring 1 (18) + νCC (19) |
| 90 | 1315 | 1264 | 61.155 | νCC ring 3 (11) + δHCC ring 3 (46) |
| 89 | 1290 | 1241 | 8.991 | νCC Δ (17) + δHCC Δ (12) + δHCC ring 1 (27) |
| 88 | 1285 | 1236 | 14.092 | νCC Δ (17) + δHCC ring 1 (28) |
| 87 | 1270 | 1222 | 119.66 | νCC ring 2 (37) + νNC (10) |
| 86 | 1264 | 1215 | 117.24 | νCC ring 3 (51) + δHNC (18) + δHCC ring R3 (10) |
| 85 | 1229 | 1182 | 6.642 | νCC (15) + δHCC Δ (13) + δCCC ring 1 (15) |
| 84 | 1222 | 1176 | 12.894 | νCC ring 3 (43) + δHCC ring 3 (19) |
| 83 | 1216 | 1169 | 17.488 | νCC ring 2 (16) + δHCC ring 2 (62) |
| 82 | 1190 | 1143 | 0.337 | νCC ring 3 (10) + δHCC ring 3 (79) |
| 81 | 1176 | 1130 | 2.746 | νCC ring 1 (11) + δHCC ring 1 (51) |
| 80 | 1151 | 1106 | 27.913 | δHCC ring 3 (12) + δHCC ring 2 (12) + δHCC ring 3 (13) |
| 79 | 1148 | 1104 | 6.561 | νCC ring 2 (12) + δHCC ring 2 (34) |
| 78 | 1143 | 1099 | 38.136 | δHCC ring 2 (14) + τHCCC Δ (12) + τHCCC Δ (16) |
| 77 | 1135 | 1091 | 14.121 | νCC ring 1 (46) + δHCC ring 1 (23) |
| 76 | 1118 | 1075 | 18.110 | νNC (26) + δCCC R3 (12) |
| 75 | 1092 | 1048 | 5.129 | τH CCC Δ (49) |
| 74 | 1077 | 1036 | 1.458 | νCC ring 3 (50) + δHCC ring 3 (12) |
| 73 | 1071 | 1027 | 3.629 | δCH3 (16) + τCCCH3 (52) |
| 72 | 1066 | 1022 | 38.567 | τHCCC Δ (53) + τHCCC Δ (12) |
| 71 | 1033 | 993 | 30.526 | δCCC ring 2 (66) |
| 70 | 1025 | 986 | 32.326 | νCC ring 1 (11) + δCCC ring 1 (73) |
| 69 | 1022 | 982 | 13.09 | νCC Δ (12) + δCCC Δ (24) + τHCCC Δ (25) |
| 68 | 1017 | 976 | 2.782 | δCH3 (11) + δCCC ring 3 (12) + τCCCH3 (48) |
| 67 | 1001 | 962 | 1.985 | τHCCC ring 2 (78) |
| 66 | 984 | 944 | 0.064 | τHCCC ring 3 (77) |
| 65 | 982 | 942 | 1.188 | τHCCC (61) + τCCCC (13) |
| 64 | 969 | 931 | 1.460 | δHCC (14) + τHCCC ring 2 (13) + τHCCC ring 1 (10) |
| 63 | 967 | 928 | 0.318 | τHCCC ring 2 (60) + τCCCC (13) |
| 62 | 947 | 909 | 2.779 | τHCCC ring 3 (68) τCCCN (10) |
| 61 | 919 | 883 | 3.003 | δOCN (12) + δCNC (10) τ+ HCCC ring 3 (15) |
| 60 | 917 | 882 | 21.721 | νCC Δ (21) + δHCC Δ (11) + τHCCC (31) |
| 59 | 914 | 880 | 2.327 | νCC ring 2 (14) + δHCC ring 2 (12) + τHCCC (39) |
| 58 | 888 | 853 | 2.751 | δHCC Δ (13) + τHCCC ring 2 (24) |
| 57 | 871 | 835 | 2.681 | τHCCC ring 3 (78) |
| 56 | 865 | 832 | 11.09 | νCC Δ (10) + δCCC ring 3 (19) + τHCCC ring 2 (10) |
| 55 | 860 | 826 | 5.697 | τHCCC ring 2 (32) + τHCCC ring 3 (29) |
| 54 | 850 | 815 | 9.570 | τHCCC ring 1 (79) |
| 53 | 840 | 807 | 32.519 | τHCCC ring 2 (42) |
| 52 | 814 | 782 | 20.861 | νCC Δ (10) + δCCC ring 1 (17) |
| 51 | 780 | 750 | 28.59 | νCC (15) + τHCCC ring 3 (22) |
| 50 | 769 | 739 | 21.182 | τCCCC ring 2 (13) + ωONCC (29) |
| 49 | 763 | 732 | 25.49 | τHCCC ring 3 (60) |
| 48 | 746 | 717 | 7.065 | τHCCC ring 1 (10) + ωCCCC (42) |
| 47 | 735 | 707 | 2.38 | δCCC ring 2 + (15) τCCCC ring 3 (–13) |
| 46 | 721 | 693 | 2.41 | ωCCCC ring 2 (11) + τCCCC ring 3 (34) |
| 45 | 707 | 680 | 16.799 | τCCCC ring 2 (30) + ωONCC (26) |
| 44 | 679 | 653 | 14.69 | δCCC ring 1 (50) |
| 43 | 676 | 649 | 13.30 | δCCC ring 1 (14) + ωFCCC (15) |
| 42 | 649 | 624 | 0.44 | νCC (10) + δCCC ring 2 (78) |
| 41 | 624 | 599 | 10.24 | δCCC ring 3 (44) |
| 40 | 614 | 590 | 4.469 | δCCC ring 3 (12) |
| 39 | 582 | 558 | 10.24 | ωCCCC ring 2 (22) + ωCCCC ring 1 (10) |
| 38 | 569 | 546 | 18.887 | τCCCC (–12) + τHNCC (–13) |
| 37 | 556 | 533 | 2.953 | δCCC (13) + τCCCC ring 3 (25) |
| 36 | 544 | 517 | 49.335 | νCC (12) + δCCC ring 3 (–26) + τHNCC (46) |
| 35 | 535 | 511 | 4.433 | τHNCC (59) + τCCCC ring 3 (25) |
| 34 | 493 | 473 | 6.076 | δFCC (23) |
| 33 | 473 | 455 | 5.459 | ωCCCC ring 2 (12) |
| 32 | 464 | 445 | 1.617 | τCCCC (49) |
| 31 | 458 | 440 | 8.098 | τCCCC ring 1 (27) + τCCCC ring 2 (19) |
| 30 | 445 | 428 | 1.711 | δCCC (12) + δCCN (18) |
| 29 | 441 | 423 | 3.507 | τCCCC ring 1 (17) |
| 28 | 420 | 404 | 1.652 | τHCCC ring 2 (11) + τHCCC ring 2 (13) + τCCCC ring 2 (64) |
| 27 | 406 | 389 | 0.321 | δCCC (13) |
| 26 | 403 | 387 | 4.697 | δCCBr (50) |
| 25 | 365 | 351 | 3.906 | δFCC (16) |
| 24 | 350 | 336 | 11.990 | νCC (39) |
| 23 | 330 | 316 | 0.5234 | δCCC (37) |
| 22 | 311 | 299 | 0.974 | νCC (11) + ωBrCCC (17) |
| 21 | 296 | 284 | 8.921 | δOCN (12) + δCCC (11) + τCCCN (25) |
| 20 | 277 | 265 | 3.050 | τCCCN (25) |
| 19 | 256 | 245 | 1.391 | ωCCCC (11) + ωCCCC (21) |
| 18 | 231 | 219 | 4.296 | δCCC (11) |
| 16 | 194 | 183 | 0.349 | νBrC (14) δCCC (19) |
| 15 | 186 | 168 | 1.42 | νBrC (14) + δCCC (19) |
| 14 | 172 | 163 | 1.027 | δNCC (14) + δCCC (10) + τHCCC (21) |
| 13 | 154 | 147 | 0.453 | τHCCC (10) + ωCCCC (13) + ωBrCCC (12) |
| 12 | 133 | 127 | 0.344 | ωCCCC (22) + ωBrCCC (12) |
| 11 | 119 | 112 | 1.968 | δBrCC (76) |
| 10 | 108 | 102 | 1.626 | ωCCCC (10) + τCNCC (40) |
| 9 | 104.6 | 100 | 1.851 | δCCC (13) + τCCCC (12) + τCNCC (20) |
| 8 | 83 | 79 | 0.237 | τCCCC (34) |
| 7 | 60.82 | 56 | 0.044 | δCCC (38) |
| 6 | 52.01 | 49 | 0.113 | τCNCC (53) |
| 5 | 48. | 46 | 0.118 | δCCC (11) + δCCC (17) + δCNC (12) + ωCCCC (15) |
| 4 | 34 | 30 | 0.144 | δCCC (12) + δCNC (33) |
| 3 | 20.93 | 20 | 0.090 | τCNCC (46) |
| 2 | 19.35 | 13 | 0.638 | δCNC (12) + τCCCC (55) |
| 1 | 14.54 | 11 | 0.0595 | τCNCC (30) + τCNCC (12) |
| ν: stretching, δ: bending, τ: twisting, γ: out of plane bending, s: symmetric, as: asymmetric | ||||
下载: 导出CSV
Table 4. Calculated Energy Values for (DBFB) Using B3LYP/6-31G(d, p)
| Parameters | Values (eV) |
| EHOMO | −6.226 |
| ELUMO | −1.395 |
| Gap (ΔE) | 4.832 |
| Electronegativity (χ) | 3.811 |
| Chemical potential (μ) | −3.811 |
| Chemical hardness (η) | 4.831 |
| Chemical softness (S) | 0.207 |
| Electrophilicity index (ω) | 1.503 |
| Additional electronic charge (ΔNmax) | 0.163 |
下载: 导出CSV
Table 5. Mulliken Atomic Charges for DBFB
| Atoms | Mulliken atomic charges | Atoms | Mulliken atomic charges | |||
| DFT | HF | DFT | HF | |||
| Br(1) | –0.094459 | –0.075057 | C(16) | 0.553734 | 0.795130 | |
| Br(2) | –0.093303 | –0.073689 | C(21) | –0.098879 | –0.159134 | |
| N(1) | –0.639660 | –0.808507 | C(22) | –0.10338 | –0.142897 | |
| F(1) | –0.301539 | –0.400608 | C(23) | –0.385374 | –0.346166 | |
| O(1) | –0.503772 | –0.594833 | C(19) | –0.139060 | –0.166692 | |
| C(1) | 0.039286 | –0.027181 | C(20) | –0.074642 | –0.140331 | |
| C(2) | 0.036221 | –0.021984 | H(4) | 0.261137 | 0.309928 | |
| C(3) | –0.139351 | –0.122195 | H(3) | 0.116343 | 0.189077 | |
| C(4) | 0.196518 | 0.059095 | H(5) | 0.094871 | 0.164301 | |
| C(5) | –0.131284 | –0.157487 | H(6) | 0.116593 | 0.185202 | |
| C(6) | –0.088779 | –0.111020 | H(7) | 0.111783 | 0.157378 | |
| C(7) | –0.203012 | –0.214280 | H(9B) | 0.141130 | 0.165981 | |
| C(8) | 0.297273 | 0.445493 | H(9A) | 0.126005 | 0.149713 | |
| C(9) | –0.248240 | –0.283870 | H(15) | 0.112567 | 0.189030 | |
| C(10) | 0.116477 | –0.032461 | H(14) | 0.124193 | 0.201208 | |
| C(15) | –0.128951 | –0.154107 | H(11) | 0.086824 | 0.15351 | |
| C(14) | –0.100554 | –0.111773 | H(20) | 0.084919 | 0.151090 | |
| C(13) | 0.038415 | –0.151646 | H(21) | 0.086260 | 0.151493 | |
| C(12) | –0.132461 | –0.142267 | H(22) | 0.079983 | 0.151043 | |
| C(11) | –0.146731 | –0.176673 | H(23) | 0.109964 | 0.12170 | |
| H(19) | 0.082299 | 0.148166 | H(23B) | 0.162564 | 0.171392 | |
| C(17) | 0.232932 | 0.247133 | H(23A) | 0.100062 | 0.115260 | |
| C(18) | 0.151961 | 0.026484 | H(12) | 0.093125 | 0.166038 | |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们







 下载:
下载: