引用本文:
杨新, 杨文, 邓颖颍, 杨定乔. 过渡金属催化二环烯烃加成反应的研究进展[J]. 有机化学,
2017, 37(10): 2512-2525.
doi:
10.6023/cjoc201604029

Citation: Yang Xin, Yang Wen, Deng Yingying, Yang Dingqiao. Progress in Transition-Metal Catalyzed Bicyclic Olefins Addition Reaction[J]. Chinese Journal of Organic Chemistry, 2017, 37(10): 2512-2525. doi: 10.6023/cjoc201604029
Citation: Yang Xin, Yang Wen, Deng Yingying, Yang Dingqiao. Progress in Transition-Metal Catalyzed Bicyclic Olefins Addition Reaction[J]. Chinese Journal of Organic Chemistry, 2017, 37(10): 2512-2525. doi: 10.6023/cjoc201604029
过渡金属催化二环烯烃加成反应的研究进展
English
Progress in Transition-Metal Catalyzed Bicyclic Olefins Addition Reaction
Abstract:
In this paper, the recent research progress in transition-metal catalyzed bicyclic olefin addition reactions is reviewed, mainly including Ru, Rh, Ir, Pd, Cu, Co, Ni and Fe etc. Moreover, the possible mechanisms of some parts of addition reactions are also discussed.
-
Key words:
- transition-metal
- / bicyclic olefins
- / addition reaction
- / research progress
-
过渡元素是指元素周期表中d区的一系列金属元素, 又称过渡金属, 由于空的d轨道的存在, 过渡金属很容易与一些配体络合形成均相催化剂.该类型催化剂活性中心比较专一, 具有较高的对映选择性和区域选择性, 副反应少, 反应活化能低, 反应速率快, 易于用光谱、波谱、同位素示踪等方法来研究催化剂的作用等特点, 因此得到越来越多的化学家的青睐[1~3].过渡金属络合物的配位基团可以适当地选择, 使金属原子周围具有特定的电子与空间性质, 从而使它仅对某一特定反应具有高度的化学选择性和立体选择性, 同时它的可调变性往往又可用于不同类型底物的催化.
根据含不同原子(C、N和O)桥环二环烯烃, 可分为氧(氮)杂二环烯烃和非氧(氮杂)二环烯烃.它们很容易由不同类型苯炔或乙烯、环戊二烯、吡咯和呋喃通过Diels-Alder环加成反应制得.二环烯烃除了在过渡金属不对称催化开环反应有广泛的应用外, 在过渡金属催化加成反应方面也具有广阔的应用前景.
加成反应可分为环加成、亲核加成和亲电加成等几类.环加成反应(Cycloaddition Reaction)是合成环状分子化合物的一种较好的方法之一, 其主要特点是将不饱和链状化合物直接转变为环状化合物[4].如[2+2]环加成形成四元环结构, 许多药物和天然产物分子中都含有四元碳环结构单元.亲核加成是形成碳-碳键的一种重要方法, 而碳-碳键是构建有机化合物碳骨架的基础, 所以碳-碳键形成方法的研究具有重要的现实意义和科学价值[5].
我们课题组主要从事过渡金属催化氧(氮)杂二环烯烃不对称开环反应, 已经发表了过渡金属不对称催化氮杂二环烯烃[6]或氧杂二环烯烃[7]开环反应研究进展的综述, 还发表了铑[8]、钌[9]、铱[10]、钯[11]、镍[12]和铂[13]等过渡金属催化环加成反应研究进展的综述.其他课题组相关的综述报道有二环烯烃促进的钯催化多米诺Catellani反应研究进展[14]和基于导向策略的钯催化间位C(sp2)—H键活化反应的研究进展[15], 但近年来对二环烯烃加成反应的研究进展还没有报道.本文将主要综述过渡金属催化含氮(氧)杂原子或不含杂原子二环烯烃与炔烃的环加成反应, 以及与芳烃、末端炔烃和苯硫酚等亲核加成反应的研究进展.重点讨论钌、铱、铑、钴、钯、镍、铜和铁等不同的催化剂催化加成反应, 并对可能的部分加成反应机理进行了讨论.
1 钌催化二环烯烃的加成反应
2003年, Tam等[16]报道了Cp*Ru(COD)Cl催化二环烯烃1与手性取代炔丙基醇2发生[2+2]环加成反应(Eq. 1).环加成产物3和4, 其主要立体构型为anti-exo, 而且该反应具有很高的立体化学选择性, 能获得中等产率的加成产物3和4.当二环烯烃取代基X=H, Y=H或t-BuO时, 与手性丙炔醇的羧酸酯反应, 产率上升到90%, 加成产物3和4的非对映选择性dr值比值为84:16.
2006年, Tam等[17]基于Eq. 1的基础上, 实现了二环烯烃5与炔6的[2+2]环加成反应(Eq. 2).当底物5 X=N-Boc时, 与炔丙醇6f反应, 仅得到一种环丙烷化产物8, 产率为72%.这是首次通过Cp*Ru(COD)Cl催化体系, 得到具有高立体选择性的环丙烷加成产物.研究表明:不同的催化剂对加成产物7与8的产率有很大影响, 当催化剂为Cp*Ru(COD)I时, 主要得到含环丁烯结构的加成产物7, 加成产物7与8产率比值为87:13.当催化剂为Cp*Ru(COD)Cl时, 主要得到环丙烷化的产物8, 加成产物7和8产率比值为31:69.当催化剂为Cp*Ru(COD)Br时, 加成产物7和8产率比值为49:51.
2006年, Tenaglia等[18]报道了一种新型的CpRuCl-(PPh3)2催化剂, 相比于Eq. 2中的Cp*Ru(COD)Cl催化剂, 富电子的环戊二烯基钌配合物CpRuCl(PPh3)2在催化二环烯烃5与末端炔酸酯9的加成反应时, 只得到[2+1]环加成产物(1, 2, 3-三取代的环丙烷10), 而不会得到[2+2]环加成产物, 在该反应中环丙烷化产物10产率高达98%, 相比于Eq. 2产率明显提高(Eq. 3).
2007年, Burton等[19]报道了以Cp*Ru(COD)Cl为催化剂, 以THF为溶剂, 实现了二环烯烃11与3-苯基丙炔酸乙酯12的[2+2]环加成反应(Eq. 4).如果使用取代氮杂二环烯烃11 (R1=H, X=NBoc)作为底物时, 环加成产物13产率为88%.研究表明:二环烯烃11上杂原子的类型以及双环桥头碳上有取代基, 对加成反应速率有很大影响(表 1).当二环烯烃X=O时, 可以显著提高加成反应的速率, 这是由于二环烯烃11上的氧原子与Ru配位能力更强, 能够在催化循环中降低反应的活化能, 从而加快反应速率.而当二环烯烃11 R1有取代基存在时, 会阻碍二环烯烃11双键与催化剂Ru的配位, 从而降低反应速率.
 表 1
不同取代基的底物11对[2+2]环加成反应相对速率的影响
Table 1.
Effects of different substituent substrate 11 on the [2+2] cycloaddition reaction relative rate
表 1
不同取代基的底物11对[2+2]环加成反应相对速率的影响
Table 1.
Effects of different substituent substrate 11 on the [2+2] cycloaddition reaction relative rate
Entry 11 X R1 R2 相对速率 1 11a O H COOMe 162 2 11b O H Ph 100 3 11c O COOMe COOMe 25 4 11d CH2(a) H H 13 5 11e N-Boc H Ph 7.5 6 11f CH2(a) H Ph 7.2 7 11g O H CH2OCH3 4 8 11h CH2(b) H H 1 2008年, Tam等[20]报道了在Cp*Ru(COD)Cl为催化剂的条件下, 实现了2, 5-降冰片二烯14与不同取代基炔烃15的[2+2]环加成反应(Eq. 5).当X=N(CO2Me)R'时, [2+2]环加成产物16产率高达97%.
2009年, Tam等[21]又以Cp*Ru(COD)Cl为催化剂, 首次报道了二环烯烃17与缺电子O, O-二烷基炔基磷酸酯18 [2+2]环加成反应(Eq. 6), 加成产物19的产率高达96%.由于缺电子的O, O-二烷基炔基磷酸酯18相比炔基卤化物、硫化物、氨化物具有较低的反应活性, 因此该反应需要较高的温度和较长的反应时间, 但由于生成的目标产物O, O-二烷基烯基膦酸酯19具有一定的生物活性, 在代谢过程中, 作为抗癌和抗病毒的药物, 也是一种抗菌和抗真菌的化合物, 因此具有一定的科学意义和潜在的应用价值.
2013年, Jack等[22]尝试了以Cp*Ru(COD)Cl为催化剂, 相比反应(Eq. 6), 在不需要O, O-二烷基炔基磷酸酯18的情况下, 实现了氧杂二环烯烃20的[2+2]环双聚反应(Eq. 7).加成产物21为syn-双聚体, 产率达到66%, 该反应具有很高的区域选择性.
他们还提出了该反应可能的反应机理(Scheme 1):首先Cp*Ru(COD)Cl解离失去配体COD A, 生成Cp*RuCl B, 然后钌配合物B在氧杂二环烯烃20 C2和C3之间位置上配位, 生成配合物C; 随后通过Ru氧化插入环化作用, 生成中间体Ru环戊烷D, D再经过还原消除过程生成二聚氧杂二环烯烃21, 并再生Ru催化剂.催化剂的优先配位构型为exo, 该构型在空间上更有利于催化反应.
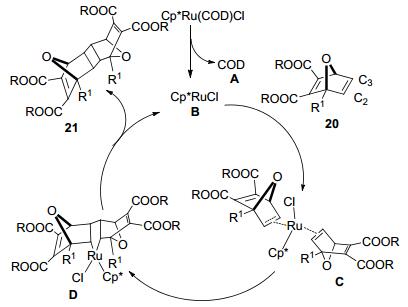 图式 1
氧杂二环烯烃20环加成反应可能的反应机理
Scheme1.
Plausible reaction mechanism for oxabicyclic olefin 20 cycloaddition
图式 1
氧杂二环烯烃20环加成反应可能的反应机理
Scheme1.
Plausible reaction mechanism for oxabicyclic olefin 20 cycloaddition
2014年, Tam等[23]报道了以5~10 mol% [Cp*Ru-(COD)Cl]为催化剂, 以THF为溶剂, 室温下反应70 h, 手性酰基樟脑磺酰胺取代的炔烃22与杂原子二环烯烃23的[2+2]发生环加成反应(Eq. 8).加成产物24主要立体构型为exo, ee值高达99%, 产率高达99%.
2015年, Bolm等[24]采用芳环上C—H键活化的方法, 报道了钌([RuCl2(p-cymene)]2)催化氧(氮)杂二环烯烃5与2-苯基吡啶25发生氢化芳基化反应(Eq. 9), 获得相应的加成产物26, 产率为94%.此反应需通入氧气才能获得高产率, 若反应在空气中进行, 产率为35%, 若反应在氩气中进行, 产率仅为5%.带有吸电子基或推电子基的2-苯基吡啶25与氧(氮)杂二环烯烃5反应, 均能在二环烯烃双键上生成加成产物26, 并具有良好的产率(65%~94%).而且在没有添加剂的情况下, 更有利于反应进行.
Bolm等提出了可能的反应机理(Scheme 2), 首先[RuⅡ]催化剂插入(可逆地)到25中2-位芳基C—H键中, 2-苯基吡啶25失去一个质子形成钌环27, 然后与7-氧杂苯并降冰片烯5a加成反应生成新的Ru配合物28, 再通过28的质子化生成产物26和起始催化剂[RuⅡ], 然后[RuⅡ]重新进入催化循环体系.这虽然解释了氢化芳基化产物的生成, 但对于该反应机理, 论文作者仍有不明确之处:一是不需要添加剂活化催化剂; 二是为什么通氧气对反应有显著的活化效果.
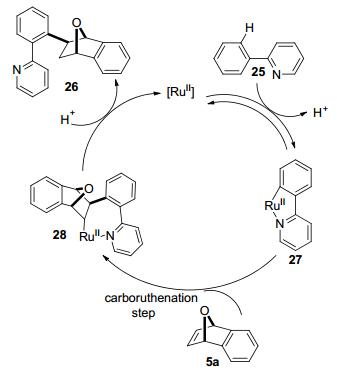 图式 2
7-氧杂苯并降冰片烯5a与2-苯基吡啶25可能的反应机理
Scheme2.
Plausible reaction mechanism for 7-oxabenzonor-bornadiene 5a with 2-phenylpyridine 25
图式 2
7-氧杂苯并降冰片烯5a与2-苯基吡啶25可能的反应机理
Scheme2.
Plausible reaction mechanism for 7-oxabenzonor-bornadiene 5a with 2-phenylpyridine 25
近年来, 已经报道了一些实用的方法催化二环烯烃30快速合成七环-[6.6.0.0.0.0.0.0]十四烷31 (heptacyclo-[6.6.0.0.0.0.0.0]tetradecanes, 简称HCTD)和各种新的7, 12-双取代HCTDs (Eq. 10).相比之前的方法, 2016年, Dong等[25]选择使用市售的一般规格催化剂, 而不是精密准确浓度的催化剂, 并用此催化剂催化反应, 就可以使目标产物31具有良好的产率(78%).此外, 这是首次实现四环烷烃29环加成反应, 加成产物31 HCTD具有良好的产率.而且HCTDs的衍生物31可以有效地制备一系列同型和异型双功能化骨架, 它在生物和材料方面具有潜在的应用价值.
2 铱催化二环烯烃的加成反应
2010年, Shao等[26]首次报道了取代氧杂二环烯烃5与末端炔烃32的[2+2]环加成反应(Eq. 11).研究表明:铱催化[2+2]环加成反应能一步生成四个立体中心的加成产物, 环加成产物33产率上升为81%, ee值上升为99%.
2012年, Kwong等[27]首次报道了以[Ir(COD)Cl]2为催化剂, (R)-synphos 37为手性配体, 实现了二环烯烃14与芳基末端炔烃35的不对称氢炔化反应(Eq. 12).加成产物36具有很高的对映选择性, ee值高达97%, 产率高达92%.
2013年, Fan和Chan等[28]仍以[Ir(COD)Cl]2与(R)-synphos (37)手性配体形成催化剂, 催化不同取代基的氧杂二环烯烃5与末端炔烃32的加成反应(Eq. 13).当R1=R2=H, R=Ph时, 加成产物38产率为63%, ee值为77%.当把产物经过简单的重结晶后, ee值能提高到96%.通常, 该反应具有良好的官能团耐受性, 能够得到具有良好产率(51%~90%)和对映选择性(62%~78% ee)的加成产物, 但当末端炔烃32中R=4-CH3OC6H4时, 取代氧杂二环烯烃5的取代基和取代基位置对反应的产率和对映选择性有很大影响(表 2).研究表明, 带有吸电子基团的底物5更有利于对映选择性; 当取代基靠近双键的位置时, 不利于反应的进行, 当取代基在双环桥头上则会阻止反应的进行.这是因为立体位阻的结果.
表 2
不同取代基底物5对加成反应时间、产率和ee值的影响
Table 2.
Effects of different substituted substrate 5 on the addition reaction time, yield and ee values
Entry R1 R2 Time/h Yield/% ee/% 1 OMe H 19 92 67 2 H Me 19 81 75 3 H Br 19 84 85 4 H OMe 60 66 76 5 H 1, 4-Dioxine 65 75 73 6 H 1, 3-Dioxole 46 70 72 2013年, Fan等[29]以[Ir(COD)Cl]2和(R)-xylyl-phanephos (34)络合形成的催化体系, 催化氧(氮)杂二环烯烃39与苯乙炔40 [2+2]环加成反应(Eq. 14).结果表明, 底物39上的杂原子对反应的对映选择性具有很大的影响.当X=O, N时, 目标产物总是能获得较高的对映体过量值(90%~90% ee).例如, 当R1=R2=R3=H, X=O时, 目标产物41的ee值高达99%;当X=NBs时, 目标产物41的ee值高达95%.而当X=CH2时, 催化剂对反应对映选择性的控制能力明显降低.作者还发现; 当底物39苯环上带有取代基时, 反应的对映选择性略微降低, 若双环桥头上连有取代基R1时, 由于立体障碍的作用, 将会阻碍反应的进行.
2014年, Fan等[30]以[Ir(COD)Cl]2为催化剂, (R)-xylyl-phanephos (34)为手性配体, 报道了苯乙炔(40)对外消旋(±)-氧杂二环烯烃42的[2+2]环加成反应的动力学拆分的研究(Eq. 15).当R=CH3时, 环加成产物43的产率为36%, ee值高达99.9%; (-)-氧杂二环烯烃44经拆分后回收率为45%, ee值高达97%.
2015年, Yamamoto等[31]以[Ir(COD)2](BAr4F)和(R, R)-S-Me-BIPAM (45)手性配体络合形成的配合物作为催化剂, 催化二环烯烃47与邻位C―H键活化的苯乙酮或苯甲酰胺类衍生物46发生加成反应(Eq. 16).该加成反应具有良好的对映选择性.值得注意的是, 二环烯烃47与N, N-二烷基苯甲酰胺的反应, 加成产物48产率高达97%, ee值高达99%.
2015年, Fan和Kwong等[32]报道了铱与配体(R)-synphos (37)络合形成的催化体系, 催化取代二环烯烃5与末端炔烃32的加成反应(Eq. 17).该加成反应具有较高的产率(98%)和对映体过量值(99% ee).当催化剂为铑时, 产率只有7%.当R1, R2为不同取代基时, 对目标产物49的ee值影响不是很大, 而且这是首次实现氮杂二环烯烃与末端炔烃的加成反应.
2016年, Wang等[33]以[Ir(COD)2BF4]和手性配体(S)-xyl-binap (50)络合形成的催化体系, 催化取代氧杂二环烯烃5与芳基硫酚51不对称加成反应(Eq. 18).该催化体系避免了催化剂中毒, 并形成独特的芳基硫酚加成产物, 加成产物52的产率高达97%, ee值高达98%.而且没有发现竞争性开环产物的产生, 并通过X射线衍射晶体结构分析, 证实了加成产物52立体构型为exo.
3 铑催化二环烯烃的加成反应
2007年, Hayashi等[34]报道了铑和配体(R)-binap (53)络合形成的催化剂, 不对称催化取代氧杂二环烯烃5发生双聚反应(Eq. 19).该反应具有反应温度低、耗时短等特点, 而且目标产物54产率高达99%, ee值高达99%.
2007年, Tam等[35]在Hayashi基础上, 设计了[Rh(COD)Cl]2与手性双膦配体络合形成的催化剂, 通过一步催化反应就实现了取代氧(氮)杂二环烯烃5发生双聚反应(Eq. 20), 加成产物55为萘并[1, 2-b]呋喃环体系.作者对不同的铑(Ⅰ)催化剂、Ag(Ⅰ)盐、溶剂和双膦配体对目标产物55的产率和对映选择性的影响进行了研究, 并探索了反应局限性和适用范围.当手性双膦配体为(R)-binap (53), 添加剂为AgBF4时, 加成产物55具有较高的产率(96%)和对映体过量值(99% ee).当底物5为取代氮杂二环烯烃类似物时, 也能得到具有良好产率和高对映选择性的双聚产物.结果表明:只有底物为取代氧(氮)杂二环烯烃5, 而且双环桥头碳上没有取代基时, 才能发生环双聚反应, 作者还发现了取代氧(氮)杂二环烯烃5上不同的吸电子基团在反应中可展现出不同的反应活性.
2010年, Lautens等[36]报道了一种新型的催化剂手性N-杂环卡宾配合物Rh(IBiox[(-)-menthyl])(CO)2Cl (56).该配合物作为取代氮杂二环烯烃57不对称芳基化反应(Eq. 21)的催化剂, 使反应表现出较高的对映选择性, 56还被应用于合成地棘蛙素, 加成产物59产率为86%, ee值高于93%.
Lautens还提出了该反应可能的机理(Scheme 3), 该反应开始在无水条件下缓慢反应, 首先络合物LnRhCl转换成LnRhOH (60), 60再与芳基硼酸试剂58作用, 进行金属转移生成LnRhAr (61).然后再与取代氮杂二环烯烃57配位和Rh插入形成中间体62, 通过62 1, 4-Rh迁移生成Rh(Ⅲ)物质63, 然后64与水(1 equiv.)反应, 生成产物59并再生LnRhOH配合物60.
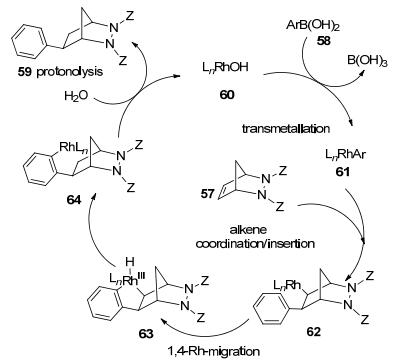 图式 3
取代氮杂二环烯烃57与芳基硼酸58反应可能的机理
Scheme3.
Plausible reaction mechanism for substituted azabicyclic olefin 57 with arylboronic acids 58
图式 3
取代氮杂二环烯烃57与芳基硼酸58反应可能的机理
Scheme3.
Plausible reaction mechanism for substituted azabicyclic olefin 57 with arylboronic acids 58
2010年, Li等[37]首次报道了以[(CO)2Rh(Ⅰ)(acac)]为催化剂, Co(Ⅲ)(acac)3为添加剂, 实现了含杂原子降冰片烯65与芳醛66的脱羰基化偶联反应(Eq. 22).该反应不需要配体就可使反应具有较好的立体选择性, 目标产物构型仅为exo型.作者还发现加入溶剂的量对反应的产率有很大影响.当加入甲苯溶剂为0.2 mL时, 偶联产物67产率为93%, 而加入甲苯溶剂为0.6 mL时, 产率仅为66%.
2013年, Fan等[38]报道了Rh前体与手性二茂铁配体(R, S)-Cy2PF-PPh2(68)配位形成的催化体系, 催化含杂原子二环烯烃5和芳基末端炔烃35不对称氢炔化反应(Eq. 23).当催化剂为RhCl3•3H2O时, 选择不同的底物均可以获得具有较高产率(99%)和对映体选择性(ee>99.9%)的加成产物69.
2014年, Bolm等[39]报道了在2.5 mol% [Cp*Rh(MeCN)3][BF4]2催化作用下, 实现了含杂原子二环烯烃5与芳基磺酰亚胺70的氢化芳基化反应(Eq. 24).加成产物71产率为70%~91%, 研究表明, 当芳基磺酰亚胺70的取代基R1为推电子基团时, 能够获得较高的产率; 当取代基为吸电子基时, 产率略微降低, 而当取代基为卤素时, 对产率影响不大.
2016年, Radhakrishnan等[40]设计了以3 mol% [RhCl2Cp*]2为催化剂, Cu(OAC)2•H2O (2 equiv.)为氧化剂, 一步实现了氮杂二环烯烃72与取代水杨醛73的氧化偶联成环反应(Eq. 25).研究表明, 该反应不需要配体, 而且氧化剂和溶剂的选择对反应有很大影响, 如选择以THF、xylene、MeOH、dioxane和toluene为溶剂时, 均无反应发生.选择以AgOAc和AgCO3为氧化剂, 仅有微量反应发生.但是选择以Cu(OAc)2•H2O为氧化剂时, 乙腈为溶剂时, 反应得到目标产物74的产率为93%.
4 钴催化二环烯烃的加成反应
2000年, Pericas等[41]通过简单的加热PuPhos-BH3 (1 equiv.)与1, 4-二氮杂双环[2.2.2]辛烷(2 equiv.)反应, 生成了游离的PuPhos配体, 然后再与二钴六羰基配合物混合, 生成的产物为非对映异构体混合物(钴四羰基配合物75与76), 当钴四羰基配合物上R=TMS时, 非对映异构体75与76比值为3:1, 在CH2Cl2/NMO/r.t.条件下, 钴四羰基配合物75与2, 5-降冰片二烯14反应, span>生成的目标产物77, 产率为93%, ee值高达97% (Eq. 26).
2012年, Hayashi等[42]报道了钴与手性配体(R, R)-QuinoxP* (78)络合形成的催化剂, 催化取代氧(氮)杂二环烯烃5与三异丙基硅乙炔79 [R3=Si(i-Pr)3]不对称加成反应(Eq. 27).不同取代基的底物5对目标产物80的产率和ee值均有影响.当R1=R2=H, X=O, R3=Si(i-Pr)3时, 加成产物80产率为91%, ee值高达99%;而当X=NBoc时, 产率明显降低为73%, ee值为96%.为了进一步研究催化循环的机理, 作者起始用氘代三异丙基硅乙炔79进行了氘标记.结果表明:末端炔烃上的氢是加在目标产物的C-3位置上.
自从Khand和Pauson使用新型的P-立体异构膦配合物发现了不对称分子间Pauson-Khand反应后, 该类型反应就一直受到化学家们的广泛关注. 2015年, Verdaguer等[43]首次用Co-双膦催化体系, 实现了2, 5-降冰片二烯14与末端炔烃35的加成反应(Eq. 28).作者还发现Co-双膦催化体系能使该反应充分进行, 而且加成产物81具有较高的对映体过量值(97% ee)和产率(97%).
2015年, Zhao等[44]在10 mol% CoBr2和10 mol% TBAI (tetra-n-butylammonium iodide)催化下, 首次实现了取代氧(氮)杂二环烯烃5与烯丙基三氟硼酸钾83的加成反应(Eq. 29).作者发现, 所有加成产物84均为syn-非对映异构体.作者认为底物5中杂原子可能与钴催化剂相互作用, 并在碳钴化步骤中, 在同一侧引导烯丙基化加成.当二环烯烃5 (X=CH2)作为底物时, 并未获得产物.总的来说, 这种简单的钴催化方法, 生成了多种新型氢化烯丙基化加成产物.当X=NBoc, R=H时, 加成产物84产率高达99%;当X=O时, 苯环上不同的取代基对目标产物84a~84i的产率有一定的影响.
5 钯催化二环烯烃的加成反应
2004年, Cheng等[45]报道了在0.025 mmol PdCl2(PPh3)2的催化下, 以乙腈为溶剂, 反应8 h, 常温下实现了二环烯烃85与86 [2+2+2]环加成反应(Eq. 30).当85为氧杂二环烯烃时(X=O), 与86a反应, 获得加成产物9, 10-二氢-菲87a, 产率为94%.当通过简单的路易斯酸介质的脱氧芳构化, 环加成产物87能被应用于多环芳香烃和有取代基的苯并三苯基烯的合成, 并且获得具有良好产率(54%~94%)的产物, 另外环加成产物87可以通过逆狄尔斯-阿尔德(Diels-Alder)反应, 为有取代基的菲和异苯并呋喃提供一种新的合成方法.
2005年, Hou等[46]报道了通过苄基噁唑啉合成的新型环钯催化剂(无膦环化钯) 88, 并使用该催化剂催化二环烯烃85或苯并二环烯烃5与碘苯89的氢苯基化反应(Eq. 31).作者发现, 当反应处于120 ℃高温时, 仅需少量催化剂(2.5×10-3mol%)就可使目标产物产物90或91产率高达97%;而当反应处于65 ℃时, 当催化剂增加到0.25 mol%时, 目标产物90或91产率可高达99%, 而且反应时间也缩短一半.
2006年, Tenaglia等[47]设计了Herrmann-Beller (赫尔曼-贝勒)钯环配合物93, 催化2, 5-降冰片二烯14与末端炔烃32的加成反应(Eq. 32).当末端炔烃32 R=1-hydroxycyclohexyl时, 目标产物92产率高达97%, 而且该加成反应具有良好的官能团耐受性.
2006年, Cheng等[48]在Pd(dba)2的催化作用下, 实现了7-氧杂苯并降冰片烯5a、对碘苯乙酮(94)和4, 5-二甲基-2-(三甲基硅基)苯基三氟甲基磺酸酯(86)的碳环化反应(Scheme 4).加成产物95为环状9, 10-二氢-菲衍生物, 产率为92%.当加成产物95以路易斯酸为介质时, 通过脱氧芳构化反应可以进一步转化为聚芳烃96, 产率高达90%.
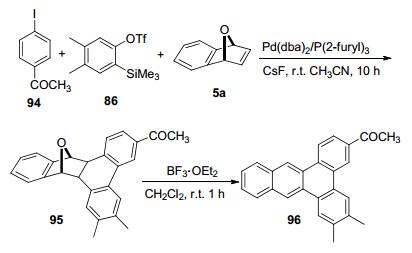 图式 4
钯催化三组分反应
Scheme4.
d-catalyzed three component reactions
图式 4
钯催化三组分反应
Scheme4.
d-catalyzed three component reactions
2008年, Liu等[49]设计了以Pd(PPh3)4为催化剂, 四丁基碘化铵(Bu4NI)为相转移催化剂, 实现了氧杂二环烯烃97、碘代烃98和末端炔烃32三组份的偶联反应(Eq. 33).作者发现, 邻位取代的芳基碘相比间位和对位取代的芳基碘, 目标产物99产率明显降低; 当碘代芳烃对位有取代基时, 推电子基有利于产率的提高, 而且该反应具有很高的立体选择性, 并通过X-ray衍射仪测定结构的立体构型, 证实了目标产物99的立体构型为exo, 产率为92%.
环化钯是催化取代氧杂二环烯烃5与末端炔烃32加成反应的高效催化剂. 2013年, Hou等[50]利用C(sp2)—Pd和C(sp3)—Pd键的环化钯配合物100和101, 实现了反应选择性的不同转换(Scheme 5).当使用C(sp3)—Pd环化钯催化剂101时, 加成产物102是主要产物, 产率为86%.当使用C(sp2)—Pd环化钯催化剂100时, 得到的主要是1, 2-cis开环产物103, 产率为85%.
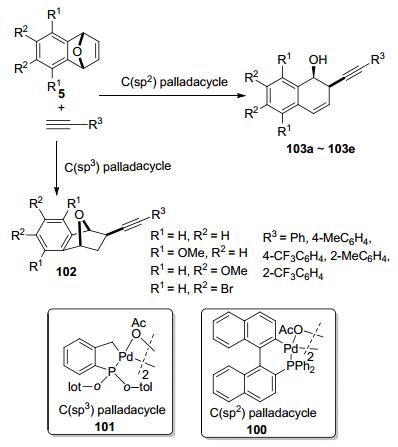 图式 5
钯催化7-氧杂苯并降冰片烯与末端炔烃的反应
Scheme5.
Pd-catalyzed 7-oxabenzonorbornadiene with terminal alkynes
图式 5
钯催化7-氧杂苯并降冰片烯与末端炔烃的反应
Scheme5.
Pd-catalyzed 7-oxabenzonorbornadiene with terminal alkynes
2014年, Wang等[51]以Pd(PPh3)4为催化剂, 催化降冰片烯65、碘代芳烃104和取代磺酰肼105三组份加成反应(Eq. 34).该反应首先发生分子间Heck反应, 随后发生烷基钯卡宾迁移插入过程, 这种转换为降冰片烯65的双官能团化提供了高效和方便的方法, 并且获得较高的产率.当碘代芳烃104中Ar=p-MeC6H4, 取代磺酰肼105中Ar1=p-AcOC6H4时, 加成产物106产率高达98%, 非对映选择性dr值为12:1.当Ar=o-MeOC6H4, Ar1=Ph时, 非对映选择性dr值虽然有所提高(20:1), 但产率明显降低为47%.
六元-七元环[4, 5, 0]杂环体系是天然产物和药物分子重要结构单元. 2014年, Hou等[52]利用环化钯催化取代氧杂二环烯烃5与末端炔烃107反应, 为制备该类型化合物提供了新方法(Eq. 35).研究表明, 当环化钯催化剂为108时, 主要为环加成产物109, 产率为72%.反应中添加剂也对环加成产物109产率有很大影响, 当反应添加剂为p-BrC6H4COOH时, 环加成产物109产率为72%, 而当添加剂为Et3N或Cs2CO3则没有任何产物生成.当环化钯催化剂为110时, 主要产物为氢炔化加成产物111, 产率为93%.
2016年, Song等[53]报道了在Pd(OAc)2与三苯基膦形成配合物作为催化剂, 催化降冰片烯14与卤代芳烃112和双(频哪醇)二硼113的芳基硼化反应(Eq. 36).该加成反应与现有的方法互补, 并且具有良好的官能团耐受性, 即使将反应的量放大到克量级, 产率也不会降低.当卤代芳烃112为4-碘甲苯时, 目标产物114产率高达93%.
6 铜催化二环烯烃的加成反应
2014年, Miura等[54]报道了CuCl与手性配体(R, R)-Ph-BPE (115)络合形成的催化体系, 催化取代氧(氮)杂二环烯烃(39)、聚甲基氢三甲基硅氧烷(116)和O-苯甲酰羟基胺117氢胺化反应(Eq. 37).目标产物118的ee值高达98%, 产率高达99%.
2015年, Hirano等[55]报道了用两种催化体系(体系A和体系B)催化二环烯烃39、O-苯甲酰基-N, N-二苄基羟胺(117)和双戊酰二硼119加成反应(Eq. 38).研究表明, 二环烯烃上不同的取代基和杂原子以及不同的催化体系条件对目标产物120的产率均有一定的影响.当选择催化体系A或B, X=O或NBoc, R1=R2=R3=H时, 目标产物120的产率高达99%.
7 镍催化二环烯烃的加成反应
2000年, Cheng等[56]报道了在[Ni(PPh3)2Cl2], PPh3和Zn粉的条件下, 以甲苯为溶剂, 实现了取代氧(氮)杂二环烯烃5与炔烃6的[2+2]环加成反应(Eq. 39).加成产物为exo-环丁烯衍生物121.当X=NCO2Me, R=H, R1=R2=Ph时, 环加成产物121产率高达98%.
2011年, Fukuzawa等[57]报道了[Ni(COD)2]与配体N-杂环卡宾122络合形成的催化剂, 催化降冰片烯65、芳香醛66和三异丙基硅烷123的偶联反应(Eq. 40).研究表明:当芳香醛66 R=4-OCH3时, 加成产物124产率高达99%.
2012年, Fukuzawa等[58]设计了[Ni(COD)2]与配体三环己基膦(PCy3)络合形成的催化剂, 催化降冰片烯65、醛66和三乙基硼125的还原偶联反应(Eq. 41).研究表明:配体PCy3和还原剂BEt3对反应有利, 而且发现多种芳香醛或脂肪醛能够发生此偶联反应, 并具有较高的非对映选择性.当R=p-MeC6H4时, 加成产物126产率为92%, dr值高于98%.
2013年, Cheng等[59]报道了在[Ni(COD)2], P(t-Bu)3, CsF, 75~85 ℃的条件下, 以甲苯和甲醇(体积比为1:3)为混合溶剂, 设计了7-氧杂苯并降冰片烯5a、有机硼酸127和炔烃6的加成反应(Eq. 44).当R1=Me, R2=Ph, R3=E-4-MeC6H4CH=CH时, 加成产物为exo-目标产物128, 产率高达95% (Eq. 42).
2016年, Cramer等[60]报道了一种新型的手性配体(N-杂环卡宾129, 简称NHC), 在NHC 129和[Ni(COD)2]络合形成的配合物催化作用下, 实现了二环烯烃14、芳香醛66和硅烷123三组份的偶联反应(Eq. 43), 偶联产物为茚满醇衍生物130.作者发现不同取代的二环烯烃14和硅烷123对偶联产物130的产率有很大影响, 但对其对映选择性影响不大.当硅烷为(i-Pr)3SiH, 二环烯烃为14b, 芳香醛66为胡椒醛时, 偶联产物130产率高达99%, 非对映选择性dr值为93:7.而当二环烯烃为14a, 芳香醛66为苯甲醛时, dr值为95.5:4.5, 但偶联产物130产率明显降低仅为25%.
8 铁催化二环烯烃的加成反应
2011年, Nakamura等[61]报道了一种新型的邻苯二芳基膦131为配体, 该配体可以有效地抑制β-杂原子的消除过程, 以1 mol% FeCl3和2 mol%配体形成配合物作为催化剂, 催化7-氧杂苯并降冰片烯(5a)与二苯基锌(132)发生碳金属化, 生成中间体133 (Scheme 6).作者发现, 不同的配体对目标产物产率影响很大.当双膦配体为131时, 主要产物为加成产物134, 产率高达95%;当配体为四甲基乙二胺(Tetramethylethylenediamine, 简称TMEDA)时, 主要产物为1, 2-cis开环产物135, 产率高达99%.
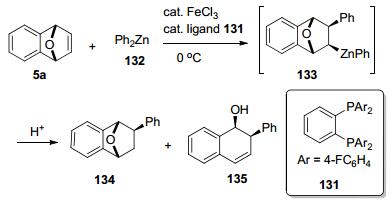 图式 6
铁催化7-氧杂苯并降冰片烯与二苯基锌的反应
Scheme6.
Fe-catalyzed 7-oxabenzonorbornadiene with diphenyl zinc
图式 6
铁催化7-氧杂苯并降冰片烯与二苯基锌的反应
Scheme6.
Fe-catalyzed 7-oxabenzonorbornadiene with diphenyl zinc
9 结论与展望
过渡金属催化二环烯烃的加成反应, 是一种构建四元碳环骨架单元和形成碳-碳键的有效方法之一.因此, 此类加成反应具有一定的科学意义和潜在的应用价值.过渡金属Ru, Ni, Cu, Rh, Ir, Co, Fe和Pd催化二环烯烃的加成反应具有较高的化学选择性和立体选择性.尽管目前已经合成出多种有效的配合物催化剂, 但没有任何一种催化剂适用于所有底物.因此, 发展高对映选择性、区域选择性和高催化活性的过渡金属配合物催化剂, 实现高效选择性的加成反应, 探索出更合适的反应条件及研究可能的催化循环反应机理仍然是将来需要研究的重点课题.
-
-
[1]
Li, C.; Zhang, H.-D.; Jiang, D.-M.; Yang, Q.-H. Chem. Commun. 2007, 6, 547.
-
[2]
Baiker, A. J. Mol. Catal. A:Chem. 1997, 115, 473. doi: 10.1016/S1381-1169(96)00352-4
-
[3]
Yang, C.-F.; Jiang, H.-Y.; Feng, J.; Fu, H.-Y.; Li, R.-X.; Chen, H.; Li, X.-J. J. Mol. Catal. A:Chem. 2009, 300, 98. doi: 10.1016/j.molcata.2008.10.041
-
[4]
陈文坤, 杨定乔, 有机化学, 2016, 36, 2075. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract345568.shtmlChen, W.-K.; Yang, D.-Q. Chin. J. Org. Chem. 2016, 36, 2075(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract345568.shtml
-
[5]
王欢, 莫海洪, 杨定乔, 有机化学, 2007, 27, 7806. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract328781.shtmlWang, H.; Mo, H.-H.; Yang, D.-Q. Chin. J. Org. Chem. 2007, 27, 7806(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract328781.shtml
-
[6]
程汉超, 梁秀丽, 李晓璐, 龙玉华, 杨定乔, 有机化学, 2012, 32, 433. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract341108.shtmlCheng, H.-C.; Liang, X.-L.; Li, X.-L.; Long, Y.-H.; Yang, D.-Q. Chin. J. Org. Chem. 2012, 32, 433(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract341108.shtml
-
[7]
韩英锋, 杨定乔, 有机化学, 2006, 26, 1613. doi: 10.3321/j.issn:0253-2786.2006.12.001Han, Y.-F.; Yang, D.-Q. Chin. J. Org. Chem. 2006, 26, 1613(in Chinese). doi: 10.3321/j.issn:0253-2786.2006.12.001
-
[8]
罗人仕, 杨定乔, 有机化学, 2007, 27, 958. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract328972.shtmlLuo, R.-S.; Yang, D.-Q. Chin. J. Org. Chem. 2007, 27, 958(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract328972.shtml
-
[9]
Hu, P.; Long, Y.-H.; Wang, H.; Mo, H.-H. Chin. J. Org. Chem. 2008, 28, 1181(in Chinese).胡萍, 龙玉华, 王辉, 莫海洪, 杨定乔, 有机化学, 2008, 28, 1181.
-
[10]
边红旭, 杨定乔, 有机化学, 2010, 30, 506. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract338794.shtmlBian, H.-X.; Yang, D.-Q. Chin. J. Org. Chem. 2010, 30, 506(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract338794.shtml
-
[11]
段泽斌, 龙玉华, 杨定乔, 有机化学, 2010, 30, 368. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract338652.shtmlDuan, Z.-B.; Long, Y.-H.; Yang, D.-Q. Chin. J. Org. Chem. 2010, 30, 368(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract338652.shtml
-
[12]
曾中一, 杨定乔, 有机化学, 2013, 33, 2131. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract342124.shtmlZeng, Z.-Y.; Yang, D.-Q. Chin. J. Org. Chem. 2013, 33, 2131(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract342124.shtml
-
[13]
程果, 杨定乔, 有机化学, 2015, 35, 2023. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract345137.shtmlCheng, G.; Yang, D.-Q. Chin. J. Org. Chem. 2015, 35, 2023(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract345137.shtml
-
[14]
朱辉, 叶长青, 陈知远, 有机化学, 2015, 35, 2291. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract345135.shtmlZhu. H.; Ye. C.-Q.; Chen, Z.-Y. Chin. J. Org. Chem. 2015, 35, 2291(in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract345135.shtml
-
[15]
袁逸之, 宋颂, 焦宁, 有机化学, 2015, 73, 1231. http://www.cnki.com.cn/Article/CJFDTOTAL-SYQY201603027.htmYuan, Y.-Z.; Song, S.; Jiao, N. Chin. J. Org. Chem. 2015, 73, 1231(in Chinese). http://www.cnki.com.cn/Article/CJFDTOTAL-SYQY201603027.htm
-
[16]
Villeneuve, K.; Jordan, R. W.; Tam, W. Synlett 2003, 2123.
-
[17]
Villeneuve, K.; Tam, W. Organometallics 2006, 25, 843. doi: 10.1021/om050780q
-
[18]
Marc, S.; Tenaglia, A. J. Org. Chem. 2006, 71, 3569. doi: 10.1021/jo060276a
-
[19]
Burton, R. R.; Tam, W. J. Org. Chem. 2007, 72, 7333. doi: 10.1021/jo701383d
-
[20]
(a) Riddell, N.; Villeneuve, K.; Tam, W. Org. Lett. 2005, 7, 3681.
(b) Villeneuve, K.; Riddell, N.; Tam, W. Tetrahedron 2006, 62, 3823.
(c) Villeneuve, K.; Riddell, N.; Jordan, R. W.; Tsui, G. C.; Tam, W. Org. Lett. 2004, 6, 4543.
(d) Allen, A.; Villeneuve, K.; Cockburn, N.; Fatila, E.; Riddell, N.; Tam, W. Eur. J. Org. Chem. 2008, 24, 4178.
(e) Riddell, N.; Tam, W. J. Org. Chem. 2006, 71, 1934. -
[21]
Cockburn, N.; Karimi, E.; Tam, W. J. Org. Chem. 2009, 74, 5762. doi: 10.1021/jo9010206
-
[22]
Jack, K.; Tam, W. J. Org. Chem. 2013, 78, 3416. doi: 10.1021/jo400104q
-
[23]
Goodreid, J.; Villeneuve, K.; Carlson, E.; Tam, W. J. Org. Chem. 2014, 79, 10002. doi: 10.1021/jo501594g
-
[24]
Cheng, H.-C.; Dong, W.-R.; Dannenberg, C. A.; Dong, S.-X.; Guo, Q.-Q.; Bolm, C. ACS Catal. 2015, 5, 2770. doi: 10.1021/acscatal.5b00258
-
[25]
Lim, H. N.; Dong, G.-B. Org. Lett. 2016, 18, 1104. doi: 10.1021/acs.orglett.6b00207
-
[26]
Fan, B.-M.; Li, X.-J.; Peng, F.-Z.; Zhang, H.-B.; Chan, A. S. C.; Shao, Z.-H. Org. Lett. 2010, 12, 304. doi: 10.1021/ol902574c
-
[27]
Fan, B.-M.; Yang, Q.-J.; Hu, J.; Fan, C.-L.; Li, S.-F.; Yu, L.; Huang, C.; Tsang, W. W.; Kwong, F. Y. Angew. Chem., Int. Ed. 2012, 51, 7821. doi: 10.1002/anie.201203107
-
[28]
Hu, J.; Yang, Q.-J.; Xu, J.-B.; Huang, C.; Fan, B.-M.; Wang, J.; Lin, C.-Y.; Bian, Z.-X.; Chan, A. S. C. Org. Biomol. Chem. 2013, 11, 814. doi: 10.1039/C2OB26775F
-
[29]
Hu, J.; Yang, Q.-J.; Yu, L.; Xu, J.-B.; Liu, S.-S.; Huang, C.; Wang, L.; Zhou, Y.-Y.; Fan, B.-M. Org. Biomol. Chem. 2013, 11, 2294. doi: 10.1039/c3ob27382b
-
[30]
Yang, Q.-J.; Yu, L.; Xu, J.-B.; Li, S.-F.; Liu, S.-S.; Chen, H.-L.; Zhou, Y.-Y.; Wang, L.; Fan, B.-M. Tetrahedron:Asymmetry 2014, 25, 957. doi: 10.1016/j.tetasy.2014.06.007
-
[31]
Shirai, T.; Yamamoto, Y. Angew. Chem., Int. Ed. 2015, 54, 9894. doi: 10.1002/anie.201504563
-
[32]
Yang, Q.-J.; Choy, P. Y.; Fan, B.-M.; Kwong, F. Y. Adv. Synth. Catal. 2015, 357, 2345. doi: 10.1002/adsc.v357.10
-
[33]
Li, S.-F.; Lu, Z.-W.; Meng, L.; Wang, J. Org. Lett. 2016, 18, 5276. doi: 10.1021/acs.orglett.6b02592
-
[34]
Nishimura, T.; Kawamoto, T.; Sasaki, K.; Tsurumaki, E.; Hayashi, T. J. Am. Chem. Soc. 2007, 129, 1492. doi: 10.1021/ja068488c
-
[35]
Allen, A.; Marquand, P. L.; Burton, L.; Villeneuve, K.; Tam, W. J. Org. Chem. 2007, 72, 7849. doi: 10.1021/jo7012884
-
[36]
Bexrud, J.; Lautens, M. Org. Lett. 2010, 12, 3160. doi: 10.1021/ol101067d
-
[37]
Yang, L.; Guo, X.-Y.; Li, C.-J. Adv. Synth. Catal. 2010, 352, 2899. doi: 10.1002/adsc.v352.17
-
[38]
Fan, B.-M.; Xu, J.-B.; Yang, Q.-J.; Li, S.-F.; Chen, H.-L.; Liu, S.-S.; Yu, Y.; Zhou, Y.-Y.; Wang, L. Org. Lett. 2013, 15, 5956. doi: 10.1021/ol402804t
-
[39]
Dong, W.-R.; Parthasarathy, K.; Cheng, Y.; Pan, F.-F.; Bolm, C. Chem.-Eur. J. 2014, 20, 15732. doi: 10.1002/chem.v20.48
-
[40]
Vijayan, A.; Baiju, T. V.; Jijy, E.; Prakash, P.; Shimi, M.; Joseph, N.; Pihko, P. M.; Varughese, S.; Radhakrishnan, K. V. Tetrahedron 2016, 72, 4007. doi: 10.1016/j.tet.2016.05.031
-
[41]
Verdaguer, X.; Moyano, A.; Pericas, M. A.; Riera, A.; Maestro, M. A.; Mahıa, J. J. Am. Chem. Soc. 2000, 122, 10242. doi: 10.1021/ja001839h
-
[42]
Sawano, T.; Ou, K.; Nishimura, T.; Hayashi, T. Chem. Commun. 2012, 48, 6106. doi: 10.1039/c2cc31880f
-
[43]
Orgue, S.; León, T.; Riera, A.; Verdaguer, X. Org. Lett. 2015, 17, 250. doi: 10.1021/ol503329g
-
[44]
Huang, Y.; Ma, C.; Lee, Y. X.; Huang, R.-Z.; Zhao, Y. Angew. Chem., Int. Ed. 2015, 54, 13696. doi: 10.1002/anie.201506003
-
[45]
Jayanth, T. T.; Jeganmohan, M.; Cheng, C. H. J. Org. Chem. 2004, 69, 8445. doi: 10.1021/jo048702k
-
[46]
Yuan, K.; Zhang, T.-K.; Hou, X.-L. J. Org. Chem. 2005, 70, 6085. doi: 10.1021/jo050491b
-
[47]
Tenaglia, A.; Giordano, L.; Buono, G. Org. Lett. 2006, 8, 4315. doi: 10.1021/ol061667e
-
[48]
Bhuvaneswari, S.; Jeganmohan, M.; Cheng, C. H. Org. Lett. 2006, 8, 5581. doi: 10.1021/ol0622918
-
[49]
Kuo, C.-J.; Cheng, S.-J.; Chang, S.-T.; Liu, C.-H. Eur. J. Org. Chem. 2008, 485.
-
[50]
Mo, D.-L.; Chen, B.; Ding, C.-H.; Dai, L.-X.; Ge, G.-C.; Hou, X.-L. Organometallics 2013, 32, 4465. doi: 10.1021/om400653x
-
[51]
Hu, F.-D.; Xia, Y.; Liu, Z.-X.; Ma, C.; Zhang, Y.; Wang, J.-B. Org. Biomol. Chem. 2014, 12, 3590. doi: 10.1039/C4OB00590B
-
[52]
Ge, G.-C.; Ding, C.-H.; Hou, X.-L. Org. Chem. Front. 2014, 1, 382. doi: 10.1039/C4QO00030G
-
[53]
Yang, K.; Song, Q. J. Org. Chem. 2016, 81, 1000. doi: 10.1021/acs.joc.5b02564
-
[54]
Miki, Y.; Hirano, K.; Satoh, T.; Miura, M. Org. Lett. 2014, 16, 1498. doi: 10.1021/ol5003219
-
[55]
Sakae, R.; Hirano, K.; Satoh, T.; Miura, M. Angew. Chem., Int. Ed. 2015, 54, 613.
-
[56]
Huang, D. J.; Rayabarapu, D. K.; Li, L. P.; Sambaiah, T.; Cheng, C. H. Chem. Eur. J. 2000, 6, 3706. doi: 10.1002/(ISSN)1521-3765
-
[57]
Ogata, K.; Atsuumi, Y.; Shimada, D.; Fukuzawa, S. I. Angew. Chem., Int. Ed. 2011, 50, 5896. doi: 10.1002/anie.v50.26
-
[58]
Ogata, K.; Toh, A.; Shimada, D.; Fukuzawa, S. I. Chem. Lett. 2012, 41, 157. doi: 10.1246/cl.2012.157
-
[59]
Mannathan, S.; Cheng, C. H. Chem. Commun. 2013, 49, 1557. doi: 10.1039/c2cc38001c
-
[60]
Ahlin, J. S. E.; Cramer, N. Org. Lett. 2016, 18, 3242. doi: 10.1021/acs.orglett.6b01492
-
[61]
Ito, S.; Itoh, T.; Nakamura, M. Angew. Chem., Int. Ed. 2011, 50, 454. doi: 10.1002/anie.201006180
-
[1]
-

图式 1 氧杂二环烯烃20环加成反应可能的反应机理
Scheme 1 Plausible reaction mechanism for oxabicyclic olefin 20 cycloaddition

图式 2 7-氧杂苯并降冰片烯5a与2-苯基吡啶25可能的反应机理
Scheme 2 Plausible reaction mechanism for 7-oxabenzonor-bornadiene 5a with 2-phenylpyridine 25

图式 3 取代氮杂二环烯烃57与芳基硼酸58反应可能的机理
Scheme 3 Plausible reaction mechanism for substituted azabicyclic olefin 57 with arylboronic acids 58

图式 5 钯催化7-氧杂苯并降冰片烯与末端炔烃的反应
Scheme 5 Pd-catalyzed 7-oxabenzonorbornadiene with terminal alkynes

图式 6 铁催化7-氧杂苯并降冰片烯与二苯基锌的反应
Scheme 6 Fe-catalyzed 7-oxabenzonorbornadiene with diphenyl zinc
表 1 不同取代基的底物11对[2+2]环加成反应相对速率的影响
Table 1. Effects of different substituent substrate 11 on the [2+2] cycloaddition reaction relative rate
Entry 11 X R1 R2 相对速率 1 11a O H COOMe 162 2 11b O H Ph 100 3 11c O COOMe COOMe 25 4 11d CH2(a) H H 13 5 11e N-Boc H Ph 7.5 6 11f CH2(a) H Ph 7.2 7 11g O H CH2OCH3 4 8 11h CH2(b) H H 1  下载: 导出CSV
下载: 导出CSV
表 2 不同取代基底物5对加成反应时间、产率和ee值的影响
Table 2. Effects of different substituted substrate 5 on the addition reaction time, yield and ee values
Entry R1 R2 Time/h Yield/% ee/% 1 OMe H 19 92 67 2 H Me 19 81 75 3 H Br 19 84 85 4 H OMe 60 66 76 5 H 1, 4-Dioxine 65 75 73 6 H 1, 3-Dioxole 46 70 72
下载: 导出CSV
-







 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 10
- 文章访问数: 4321
- HTML全文浏览量: 295
 下载:
下载:
