Citation:
Mengjiao Xie, Nadeeshani Nanayakkara, Yanbiao Liu. Pulsed electrochemistry for water decontamination: Fundamental principles and environmental application[J]. Chinese Chemical Letters,
2026, 37(4): 111669.
doi:
10.1016/j.cclet.2025.111669
Pulsed electrochemistry for water decontamination: Fundamental principles and environmental application
English
Pulsed electrochemistry for water decontamination: Fundamental principles and environmental application
College of Environmental Science and Engineering, Donghua University, Shanghai 201620, China
b.
Department of Civil Engineering, Faculty of Engineering, University of Peradeniya, Peradeniya 20400, Sri Lanka
c.
School of Environmental Science and Technology, Key Laboratory of Industrial Ecology and Environmental Engineering (Ministry of Education), Dalian University of Technology, Dalian 116024, China
* Corresponding author at: College of Environmental Science and Engineering
Received Date:
22 February 2025 Accepted Date:
01 August 2025 Revised Date:
16 June 2025 Available Online:
15 April 2026
Abstract:
Sustainable and efficient solutions are essential to address increasingly critical environmental issues, particularly in the field of pollutant removal and resource recovery. The latest research has shown that pulsed electrochemistry significantly contributes to these goals by precisely altering the local reaction environment, accelerating the reaction kinetics and decreasing the overall energy requirements. However, knowledge gaps exist in dynamic evolution mechanism of electric double layer (EDL) in this technology, and challenges remain toward fully implementation of this promising technology. In this review, the fundamentals of pulsed electrochemistry and its connection to the theoretical models of EDL are comprehensively presented. The critical parameters (e.g., duty ratio, frequency and waveform) for boosting the performance of the system are systematically discussed and the typical electrochemical reactions that occur with pulsed electrochemistry are outlined. The proposed pulsed electrochemistry methodologies tailored for environmental applications are also reviewed in detail. Finally, future opportunities and challenges of this promising but fledgling field are discussed, with the expectation that this technology offers a route to transform conventional chemical industries into cleaner and more sustainable production.
Human society is currently facing severe environmental pollution and energy crisis, and up to now, more energy efficient elimination of pollutants from water has been a significant challenge [1,2]. In recent years, interest in the use of electrochemical technologies for water treatment has increased significantly because of the need to mitigate water contamination in an energy efficient and environmentally friendly manner [3,4]. These technologies exhibit distinct advantages for water treatment in terms of their simple operation, mild reaction conditions, and minimization of secondary pollution [5]. Nearly all the studies of electrochemical water treatment have focused on optimizing the physical and chemical properties of electrodes and the rational design of system reactor to force efficient and selective electrochemical reactions [6-9]. However, the control of local reaction microenvironment remains a critical factor for boosting the electrochemical efficacy, but this issue has been largely overlooked in conventional electrochemical strategies.
In conventional electrochemical water treatment processes, direct current (DC) is customarily utilized to regulate the directional transfer of electrons, thereby effectuating the elimination of pollutants in wastewater via physical and/or chemical reactions [10,11]. However, electrode polarization tends to occur with high frequency, precipitously attenuating the overall reaction efficiency [12,13]. Moreover, within DC-driven systems, the electric double layer (EDL) gradually thickens due to the electric field restricts mass transfer of reactants from bulk solution toward the electrode/solution interface. This leads to a reduction in the transient concentration of reactants and hampers the diffusion of generated active species, consequently increasing energy consumption [14]. Conversely, pulsed electrochemistry emerges as a controllable and efficacious strategy for reaction regulation, offering better control over mass transfer kinetics and energy consumption [15-17].
Pulsed electrochemistry, characterized by periodically switching an applied potential or current to enable oxidation or reduction reactions at both electrodes, may be advantageous for generating dynamic local microenvironments. However, the apparatus, principles, and application scenarios have not been fully explored [18,19]. At the macroscopic scale, pulsed electrochemistry has a wide range of controllable reaction variables (e.g., frequency, duty ratio, and waveform function) to optimize the system compared to operating in DC mode, which can only modulate current or voltage [20]. Microscopically, electrochemical reactions usually occur at the EDL, where reactant concentrations can be intermittently renewed and regenerated by the pulsed action [21,22]. Thus, the form of EDL at the interface critically influences the thermodynamics, kinetics, and product selectivity of a variety of electrochemical processes such as the oxygen reduction reaction (ORR), the nitrate reduction reaction (NO3RR) and the carbon dioxide reduction reaction (CO2RR) [23-25]. Characterizing the structure-property relationship of the EDL has been a common topic in the field of electrochemistry [26]. Pulsed electrochemistry is also advantageous in that the configuration of the electrical pulse can be varied throughout the electrocatalytic process, resulting in a process that can be adjusted in real time to respond to specific conditions, which cannot be done with traditional systems [27]. Scientifically, the alteration of the profile of electrical pulse may seem simple, but several fundamental aspects of the mechanism and physicochemical processes need to be discerned.
To date, several insightful reviews have examined the application of pulsed electrochemistry in the field of environment. Liu et al. systematically summarized the advances on various pulsed electrochemical systems for wastewater treatment, including pulsed electrocoagulation, pulsed electro-oxidation, pulsed electro-Fenton, pulsed electrodialysis and pulsed discharge plasma [28]. Besides, a review by Wang and colleagues discussed the application of pulsed techniques on the electrodeposition synthesis of electrode materials and dynamic regulation of various electrocatalytic reactions [29]. Xi et al. provided a comprehensive overview on the pulsed technology for CO2RR on Cu-based catalysts [30]. While existing reviews tend to emphasize the application breadth or materials engineering, there is a lack of thorough examination into the underlying mechanisms that govern the pulsed-driven processes, particularly from the perspective of EDL. This oversight limits a comprehensive understanding on how the EDL dynamics influence electrochemical reaction efficiency and selectivity, thereby hindering the development of more targeted and effective pulsed electrochemistry techniques in water decontamination. Given the growing concerns regarding water contamination and the ongoing development of pulsed electrochemistry, a timely review that explores the basic mechanisms of pulsed techniques would be highly valuable for guiding future research efforts to address contamination in various environmental settings.
The primary objective of this review is to introduce the principles of the pulsed electrochemistry technique, with a focus on the EDL theory. Initially, we summarize the EDL phenomenon and the EDL model evolution. Following this, we discuss the changes in current and voltage during the EDL charging and discharging caused by the pulse. Subsequently, we delve into the specific mechanism of pulsed electrochemistry processes and discuss the essential operating factors that impacting the overall system performance. Additionally, the application of pulsed electrochemistry technology in environmental field is summarized. Finally, we conclude by outlining the challenges with the implementation of the pulsed electrochemical system and by offering a view for future developments that could overcome these difficulties.
2.
Fundamental principles
Electrochemical reactions always occur between electrolyte and electrode material, where the often-overlooked microstructure of the EDL determines the reaction kinetics in all electrochemical processes [31]. The pulsed electrochemical method has been developed into a facile and versatile system for dynamically manipulating electrochemical reactions at the electrode-electrolyte interface [32]. To fully understand the evolution of the interface during pulsed electrochemistry processes, three essential points are outlined: (ⅰ) Fundamentals of the EDL theory, (ⅱ) charging and discharging of the EDL, and (ⅲ) mechanistic insights into pulsed electrochemistry. These three points are critically important and discussed in detail in this section.
2.1
Fundamentals of the EDL theory
In any electrochemical systems, the liquid component at the solid-liquid interface consists of electrolyte, which interacts with electrostatic forces in the vicinity of the charged surface to create a layer structure called EDL near the solution’s surface [33,34]. The characteristics of this EDL region are determined by a delicate equilibrium between electrostatic and entropic driving forces [35]. Electrostatic interactions tend to promote a more compact EDL structure with a heightened local counterion density. Conversely, entropy drives the formation of thicker and more disordered EDL with lower relative interfacial potential gradients [36]. This delicate balance directly determines charge distribution, ion concentration gradient, and electric field strength on the electrode surface during electrochemical processes [37-39]. Importantly, changes within the EDL can fundamentally modulate both the adsorption of reactants and the accessibility of reactive sites, thereby dictating the reaction kinetics and selectivity [39]. While extrinsic factors (e.g., electrode surface defects, ionic species characteristics, and electrolyte composition) undeniably affect electrochemical performance, the effects of these factors are fundamentally mediated by the modulation of EDL interface. For instance, although surface defects may enhance the density of active sites, the catalytic efficacy is ultimately constrained by the intrinsically limited ion transport properties of EDL [40,41]. Similarly, the adjustment of electrolyte concentration primarily exerts the effects through modulating the EDL thickness, which subsequently reorganizes the interfacial electric field distribution [42,43]. Therefore, a deep understanding on the EDL structure plays an important role for determining the local reaction environment via pulsed electrochemistry [44,45].
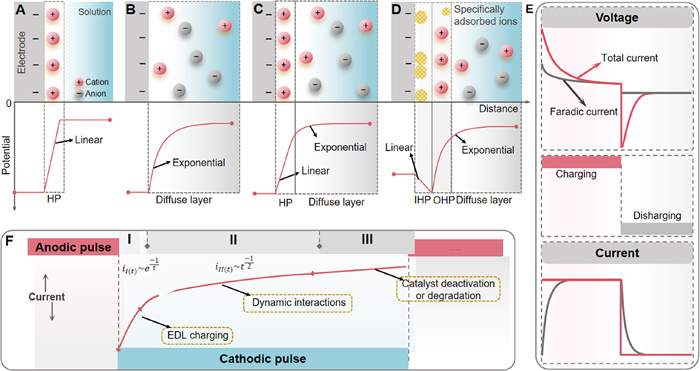
Most contemporary concepts regarding the formation of EDL largely stem from the foundational principles of classic EDL theory. The Helmholtz model, the first report on EDL model concepts [46], proposed that the large electrical capacity of the solid-liquid interface between an electrode and an electrolyte solution was caused by the presence of two charge layers that had equal but opposite electrical charges (Fig. 1A). In this model, however, the essential role of entropy in governing microscopic details of electric field screening was neglected. In contrast to Helmholtz, Gouy and Chapman (1910) described the EDL as a diffuse layer of constant thickness that accounts for the effects of ionic thermal motion (Fig. 1B) [47-49]. About a decade later, Stern bridged Helmholtz and Gouy-Chapman models by assuming that the EDL consists of two regions, i.e., an inner layer comprised of fixed ions near the surface that is inaccessible to other ionic species (referred to as the “Stern layer”), and an outer diffuse layer that accounts for heterogeneous distribution of ions (Fig. 1C) [49,50]. The Stern model intentionally discounts certain possibly important aspects of the system, including factors such as orientation and arrangement of water molecules at the surface of the charged electrode as well as the dimensions of the ions [51]. To take into account specific ions adsorption at the electrode surface, Grahame further extended the Stern model distinguishing between the two layers in the Stern model. As shown in Fig. 1D, the Stern layer can be further divided into an inner Helmholtz plane where specifically adsorbed ions reside and an outer Helmholtz plane where solvated counter ions reside [49].
Figure 1
Figure 1.
(A) Helmholtz model. (B) Gouy-Chapman model. (C) Gouy-Chapman-Stern model. (D) Grahame model. Adapted with permission [49]. Copyright 2023, Elsevier. (E) Schematic representation of current and voltages in different pulsed electrochemistry configurations. Adapted with permission [59]. Copyright 2021, Elsevier. (F) Expected current response to an applied cathodic potential. Adapted with permission [60]. Copyright 2021, Elsevier.
Based on the models described above, extensive and comprehensive efforts have been made to develop models of the EDL that take into account its solution characteristics [52,53]. Other models (e.g., Watts-Tobin [54], Schmickler [55], Badiali [56], Kornyshev [57], Price and Halley models [58]) have been successively proposed to explain different phenomena observed with the electrode materials in electrocatalysis, each with its own unique characteristics. Each of these models provides new insights into the underlying mechanisms and helps elucidate the complex electrochemistry that occurs at the interface of the solution and the electrode.
2.2
Charging and discharging of the EDL
The formation of the EDL has a significant impact on creating the local environment at the interface, where the electrochemical reactions take place. Pulsed electrochemistry enables the creation of microenvironments by modulating the EDL charging and discharging dynamics, a feature that cannot be achieved in DC systems [39,61]. Thus, it is necessary to investigate the process of EDL charging and discharging mechanisms to fully understand the fundamental processes of pulsed electrochemistry.
Once a pulse is applied, the total current (iT) supplied by the electrochemical generator comprises two components (Eqs. 1 and 2): the non-Faradaic current associated with charging and discharging of the EDL, referred to as the capacitive current (iC), and the Faradaic current (iF) that is used for electrochemical reactions [59].
iT=iC+iF
(1)
iC=−iF
(2)
The charging time is defined as the duration until iF reaches 99% of iT. If the pulse is off, i.e., the EDL is discharging (iT = 0). The energy stored within the capacitor is released through a Faraday process during the pulse-off period, in which the released current can still work. The discharging time corresponds to the period when iF decreases to between 0.010iT and 0.011iT [62].
The application of a pulse can occur in either current or voltage mode [63]. For the voltage pulse, iC approximates the charge and discharge current of an ideally polarized electrode, given by
iC=ERSexp(−tCDRS)
(3)
where E is the voltage pulse amplitude, RS is the resistance of solution, CD is the capacitance of the EDL, and t is the duration. The value of iF is estimated using Eqs. 4-6:
iF=FA(kfCO*−kbCR*)exp(H2t)erfc(Ht12)
(4)
H=kfDO12+kbDR12
(5)
erfc(x)=1−2π12∫0xe−y2dy
(6)
where A and F are the area of the electrode and the Faraday constant, respectively. The rate constants kb and kf are for oxidation and reduction reaction, respectively. CO* and CR* are the concentrations of oxidized species and reduction products in the bulk, respectively. DO and DR are the diffusion coefficients for the oxidation and reduction reaction, respectively. The term erfc(x) is the complementary error function of x [64]. As shown in Fig. 1E, when a voltage pulse is applied, the iT transitions from primarily capacitive to Faradaic as the EDL becomes completely charged. Upon returning the applied voltage to its initial value, the EDL is discharged and the current reverts to a purely capacitive current.
When a current pulse is applied, the sum of the voltages across the resistor and the capacitor (ER and EC) should be equal to the applied voltage:
E=ER+EC=iRS+qCD
(7)
Initially, the increase in E is linear with time because of the charging of the EDL, as suggested by Eq. 7 [65]. Subsequently, E is directly proportional to the square of time for a Faradaic reaction. Similar to the case of a pulse in the applied voltage, iT changes from being capacitive initially to Faradaic. During the off portion of the pulsed cycle, discharge of the EDL occurs via a Faradaic process, which causes a decay in E.
2.3
Mechanistic insights into pulsed electrochemistry
The electrochemistry associated with a pulse in voltage or current, while seemingly just a modification of the power supply mode at the macroscopic level compared to conventional constant conditions, triggers a range of significant microscopic changes. These include alterations in diffusion kinetics of reactants and products, as well as repeated potential and current transients. These transients can result in processes that are not Faradaic in nature, such as the accumulation of a surface charge, capacitive behavior, variations in the strength of the electric field of the EDL, and modifications of the structure of electrocatalysts [60].
The selectivity and reactivity of reaction processes can be altered by affecting the charging and discharging processes of EDL [66,67]. During the EDL charging process, charges rapidly accumulate on the surface of the electrode. The EDL undergoes repeated charging and discharging because of the oscillation in the applied voltage. This process effectively reduces the activation energy of the electrochemical reactions [68]. During the EDL discharging process, the change in the intensity of electric field at the EDL leads to periodic variations in ability of the electrode to adsorb and desorb reaction intermediates.
Generally, pulsed electrochemistry represents an intricate and constantly changing set of interactions at the interface between the electrode and the electrolyte [69]. At the interface, processes such as mass transport within the electrolyte, adsorption and diffusion on the electrode surface, and changes in composition and structure of the electrode collectively explicate experimental observations [70]. Notably, these processes operate cohesively at the interface rather than independently. Although it is challenging to discuss each process in isolation, their mechanism and significance can be elucidated under the electrical pulse conditions. It is important to note that fluctuations in the applied voltage or current significantly impact the interaction of these reaction processes. For example, a step change in voltage leads to a transient current response (Fig. 1F), which can be categorized into distinct regimes. In regime Ⅰ, which typically dominates for a short pulse, most of the current is directed toward charging the EDL, leaving less current available for electrocatalytic reaction. This limits the overall efficiency of the current utilization. Within regime Ⅱ, duration and voltage of the pulse potential offer a noteworthy circumstance to control the dynamic of reactants mass transport, adsorption on the electrode, and surface chemistry. However, when the applied voltage is sustained over an extended period, the system can enter into regime Ⅲ, where the electrochemical current diminishes due to deactivation or degradation of the catalyst surface [60].
Regarding the electrochemical reaction, the hydrodynamics and mass transport of reactants from the solution to the electrode need to be analyzed. Rapid reactions on the surface would reduce the quantity of reactants near the electrode, leading to solution concentration polarization [71]. The overall process is controlled by the mass transport of reactants to the active sites on the surface, corresponding to regime Ⅱ. In addition, the thickness of the boundary layer is a crucial factor in the pulsed electrochemistry process that affects the mass transport. By alternating the amplitude of the applied current or voltage, the reactants can be replenished in the EDL, increasing the concentration of reactants at the electrode surface before subsequent pulse. This provides another way to reduce the diffusion distance of reactants, thereby overcoming the mass transfer limitation. During the pulse-off period, the electrode potential may exhibit a slower decay, or an instantaneous current may be enhanced due to the discharge of the first layer of ions in the EDL, which results in the applied current or voltage being used more efficiently [37].
In terms of the reactants adsorbed on the surface of the electrode, a rapid change in the voltage can lead to a nearly complete rearrangement of the EDL and a significant change in the concentration of adsorbed reactants [72]. The resting pulse-off time (toff) allows sufficient time for pollutant molecules to diffuse from the electrode surface toward the bulk solution, creating a region of nonuniform concentration on the catalyst surface. There is ongoing diffusion of ions and pollutants toward the electrode because of gradients in concentration. The resulting dielectric relaxation at the interface between the electrode and the electrolyte reduces mass transfer limitations and helps enable reactions during the next voltage pulse [73,74].
3.
Essential operational parameters
The selection of operating parameters for pulsed electrochemistry is crucial for boosting the overall system performance. Specifically, pulse waveform, duty ratio and power frequency are viewed as key parameters [75]. In the following section, we briefly describe how the system performance correlates with these parameters.
3.1
Pulse waveform
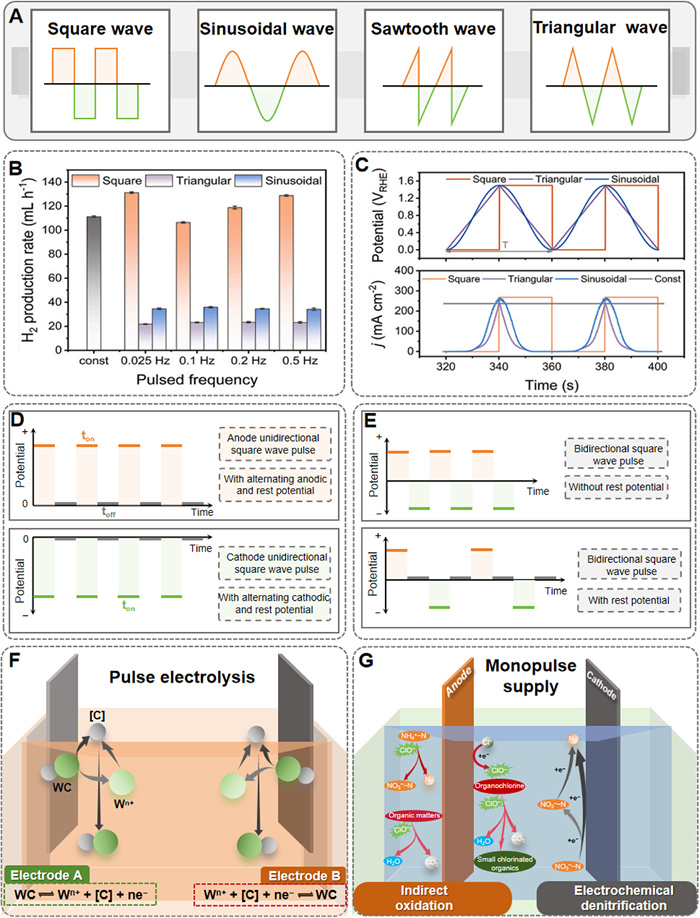
A key feature that distinguishes pulsed electrochemistry from DC operations is the waveform. The waveform of the pulse can be mainly divided into square, sinusoidal, sawtooth and triangular, as shown in Fig. 2A. The square wave is the most commonly used waveform in pulsed electrochemistry and it exhibits distinct advantages in terms of effectively enhancing the system performance [38,76]. Reports on pulsed water electrolysis demonstrated that square waveform leads to a significant hydrogen yield and the best efficiency, in contrast to triangular or sinusoidal waveforms [38,77]. The enhanced hydrogen yield demonstrated in Fig. 2B can be attributed to the distinctive characteristics of square waveforms, which enabling prolonged electrocatalytic reaction durations at elevated potentials in contrast to triangular and sinusoidal pulse waveforms [38]. This extended operational window facilitated the rapid migration of reactant ions to the electrode surface while simultaneously minimizing energy dissipation during the charge transfer [78]. Meanwhile, sinusoidal and triangular waves feature potential variations characterized by distinct periodicity and slopes (Fig. 2C). These dynamic potential alterations induce a corresponding periodic modulation of the EDL [79]. Within specific reaction systems, this may be beneficial for the generation of certain intermediate products. However, the frequent shifts in potential may also introduce complexity into the electrochemical reactions occurring at the electrode surface, thereby exerting a negative influence on the reaction selectivity.
Figure 2
Figure 2.
(A) The typical waveforms of pulsed electrochemistry. (B) Applied potential and current response curves under different pulse waveforms. (C) Hydrogen production rate versus pulse frequency under different pulse waveforms. Reprinted with permission [38]. Copyright 2024, Elsevier. (D) Unidirectional and (E) bidirectional square wave pulse with periodic switching potential. Adapted with permission [84]. Copyright 2023, Elsevier. (F) Schematic diagram of in situ pulsed synthesis of tungsten carbide. Adapted with permission [82]. Copyright 2022, Elsevier. (G) Schematic diagram of the pulsed electrochemical oxidation process for biologically treated leachate treatment. Adapted with permission [83]. Copyright 2021, American Chemical Society.
As part of the pulsed configuration, the current or voltage is cycled between two levels to generate pulses with a specific period and amplitude. Two types of square wave pulse mode with periodic switching potential are commonly employed: (ⅰ) A unidirectional square wave pulse with rest potential (Fig. 2D), and (ⅱ) a bidirectional square wave pulse with or without rest potential (Fig. 2E). Additionally, some researchers have employed varying numbers of pulse during the unidirectional or bidirectional square wave. It is worth noting that the bidirectional square wave pulse repeatedly switches the direction of the current, therefore maintaining the surface of the catalyst in a non-equilibrium state to prevent deactivation of catalyst during operation [80]. Additionally, the bidirectional square wave pulse represents a critical parameter for regulating the generation of high-efficiency products in pulsed electrochemistry applications [81]. Zhang et al. utilized pure carbon in cemented carbide to in situ regenerate high-performance tungsten carbide powder using the bidirectional pulse [82]. The positive pulse current facilitated oxidative reaction on the anode, resulting in the oxidation of tungsten carbide into carbon atoms and tungsten ions. Conversely, the negative current induced reductive reactions on the electrode, where dissolved carbon atoms and tungsten ions near the cathode were reduced into tungsten carbide powders (Fig. 2F). While this mode effectively tailored the properties of the products, the current efficiency was lower than that achieved with a unidirectional pulse due to the inherent bidirectionality of the pulse. Jiang et al. investigated electrochemical oxidation kinetics of DC, mono-pulse, and bidirectional pulse models to treat biological leachate [83]. The results showed that the mono-pulse process exhibited the best removal performance of ammonia nitrogen and other refractory organics (Fig. 2G). Therefore, the selection of appropriate square wave pulses should be tailored to meet specific water decontamination requirements.
3.2
Duty ratio
The duty ratio (r) is the primary parameter that affects the time for diffusion and reaction in the pulsed electrochemistry. The r can be viewed as the percent of time that the power supply is on and is specifically expressed as [85]:
r=tonton+toff
(8)
where ton and toff are the pulse-on and pulse-off times, respectively. Extensive reports have emerged as a key metric for evaluating the efficiency of electrochemical process, with r being widely recognized as a valuable criterion [78,86]. Sheng et al. showed that a larger r decreased the discharge intermittent time and increased the capacity per time of the discharge, thereby enhancing the number of energetic electrons and the concentration of active molecules. In addition, Ding et al. employed a pulsed voltage approach to precisely manage the 2-electron ORR and explored the effect of r on the H2O2 yield [73]. Their findings revealed that H2O2 production followed a volcano-shape trend as r increased. Intuitively, a higher pulse r meant a longer pulse-on duration, which resulted in a mass transfer limitation of reactive species during the pulse-off period, ultimately diminishing the H2O2 production. In contrast, the reaction duration necessarily decreased at lower values of r, which hindered the complete progression of the ORR. These observations underscored the critical role of r in modulating the electrochemical process and highlighted the necessity of optimizing the pulse r settings based on actual treatment conditions.
3.3
Pulse frequency
The pulse frequency (fp) refers to the reciprocal of the pulse period (T), and its value reflects the number of on and off pulse waves per second. It can be specifically expressed as:
T=ton+toff
(9)
fp=1T
(10)
As a key variable differentiating pulse from DC process, the fp directly influences reaction kinetics and system performance, thereby determining the efficiency and stability of electrochemical processes. Ma et al. carried out a systematic investigation on the effect of fp in pulse electrolysis and optimized this parameter to achieve excellent degradation performance [87]. The experimental results indicated that a higher fp can enhance the mass transfer, further improving electrochemical degradation performance. An elevated fp shortens each pulsed cycle, repeatedly interrupting electron transfer within the system. This maximizes the utilization of hydroxyl radicals and minimizes their recombination rates. Consequently, a high fp mitigates the issue of low current efficiency, yet excessively high fp can cause the pulse to regress, rendering them ineffective [88]. For example, testing the impact of different factors on the treatment of oily wastewater using pulsed electrolysis has revealed that increasing the fp causes the output current to resemble DC operation. This suppressed the mitigation of passive layers on the electrode surface and reduced the degradation of oil-containing wastewater. This phenomenon occurs because a higher fp reduces the pulse-on time, preventing the electrode-electrolyte interface from undergoing the Faraday reaction of electrochemical oxidation before the potential is cut off. As a result, the entire reaction becomes an inefficient process dominated by energy consumption for EDL charging and discharging [89]. Therefore, it is crucial to consider an appropriate fp for achieving effective operation.
4.
Environmental applications of pulsed electrochemistry
Pulsed electrochemistry technology is a cutting-edge solution to a wide range of environmental challenges, thus, offering a tailored electrode-electrolyte interface to regulate the mass transfer process and the production of valuable products by utilizing electrical pulses rather than a continuous current. A list of typical pulsed electrochemistry applications for the production of valuable materials or the removal of pollutants from water are provided in Table 1. Pulsed electrochemistry is superior in maintaining a high efficiency of an electrocatalytic reaction with a lower input of power. The following subsection will delve into the potential environmental applications of pulsed electrochemistry, with a focus on four critical areas.
Table 1
Table 1.
Summary of the parameters, performance, and contributions in typical pulse-driven electrochemical reactions.
(ⅰ) Achieving the in situ stabilization of oxygen-containing functional groups active sites; (ⅱ) Updating the interface microenvironment to promote O2 absorption and proton transfer
A 210% leap in H2O2 yield and a 74% increase in FE
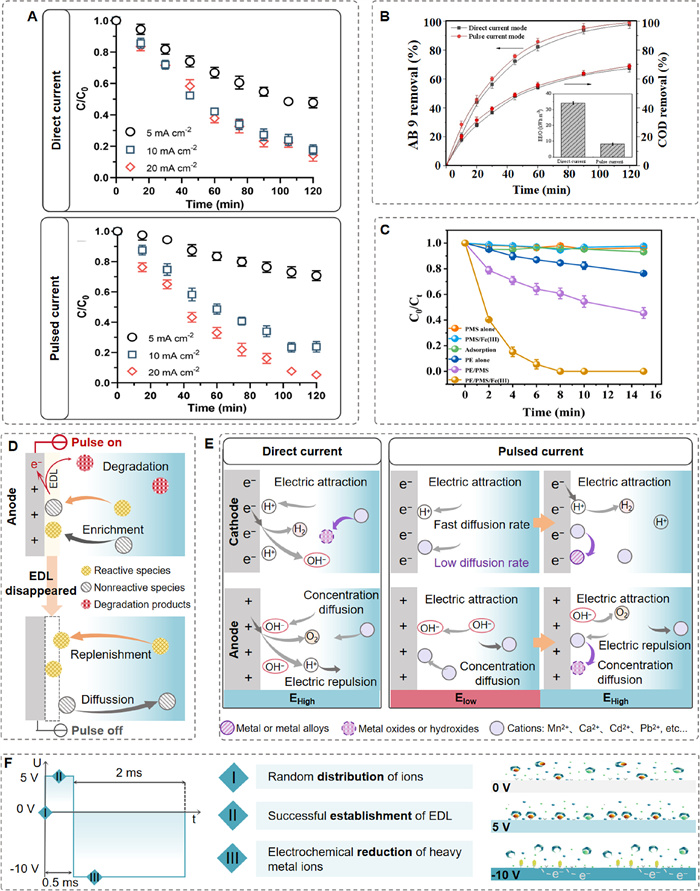
Recently, the pulsed electrochemical oxidative strategy was employed to address the process of inefficient and energy-intensive water purification, demonstrating its crucial role in reducing energy consumption and enhancing mass transfer efficiency [90-93]. For example, Pei et al. reported a 52.5% enhancement in the efficiency of phenol removal in a pulsed electrochemical oxidation process compared to a conventional DC process, as well as a 57.1% drop EPC compared to EDC (Fig. 3A) [94]. A similar pulsed oxidation process was also used to remove bio-refractory dyes and antibiotics from wastewater. Ma et al. demonstrated that pulsed electrochemical oxidation of Acid Blue 9 achieved 99.0% dye removal and 68.9% chemical oxygen demand (COD) elimination under optimized conditions, while simultaneously reducing energy consumption by 76% compared to DC mode (Fig. 3B) [87]. Furthermore, a recent investigation into the activation of peroxymonosulfate demonstrated a significant improvement in the degradation of sulfamethoxazole, achieving a 67.6% reduction in energy consumption (Fig. 3C) [95]. The improvement in degradation efficiency primarily arises from the ability of organic molecules to diffuse toward and adsorb onto the electrode under the pulse-on period, which affects the composition of the EDL [96]. The EDL dissipates quickly when the current is interrupted, resulting in a thinner diffusion layer and facilitating greater mass transfer of pollutants from the electrolyte to the electrode, thereby replenishing the local concentration changes in the EDL (Fig. 3D). This process benefits the degradation of pollutants by generating reactive oxygen species, which in turn boosts both utilization rate of reactive oxygen species and the current efficiency.
Figure 3
Figure 3.
(A) Time course of phenol oxidation during direct-current and pulsed-current electrolysis as a function of current density. Adapted with permission [94]. Copyright 2021, American Chemical Society. (B) Acid Blue 9 and COD removals at pulse and DC modes (The inset panel gives the corresponding the electrical efficiency per log order reduction value at pulse and DC modes). Adapted with permission [87]. Copyright 2021, Elsevier. (C) Comparison of performance of different oxidation processes were conducted regarding removal of sulfamethoxazole. Adapted with permission [95]. Copyright 2023, Elsevier. (D) The schematic illustration of pulse-on time and pulse-off time during pollutants degradation. Adapted with permission [94]. Copyright 2021, American Chemical Society. (E) Conversion process of heavy metal ions during pulsed electrodeposition. Adapted with permission [108]. Copyright 2024, American Chemical Society. (F) Working principle of the pulsed remediation method for heavy metal removal. Adapted with permission [109]. Copyright 2019, Springer Nature.
In addition to accelerating organic mineralization by affecting the composition of the EDL, the pulsed electrochemistry system also allows control over the selectivity of the degradation products. A recent study on biologically treated leachate demonstrated the role of pulsed electrochemistry in reducing the kinds of degradation intermediates from 53 to 11, highlighting its effectiveness in promoting the formation of reactive species by eliminating concentration polarization [83]. Despite notable improvements, achieving highly selective degradation of organic pollutant remains a challenge due to the inherent complexity of aqueous matrices. Wastewater typically contains diverse organic pollutants with differing structural and reactive activities, complicating their uniform degradation under pulsed electrochemical conditions [105,106]. Additionally, the presence of high ionic strength (e.g., Na+, Cl-) alters the electrochemical properties of the solution, disrupts the electric field distribution, and hinders pollutants diffusion and reactive species interaction, ultimately reducing the degradation efficiency [27,107].
To overcome these limitations, strategic optimization of pulse parameters offers a promising avenue. Lowering fp enables extended diffusion and reaction periods for structurally complex or low-reactivity contaminants, whereas higher fp may expedite the degradation of simpler pollutants [87]. Additionally, increasing fp can also enhance the ion migration kinetics, partially mitigating the negative effects of high ionic strength. Furthermore, coupling pulsed systems with adsorption technology offers synergistic advantages by local concentrating organic pollutants and selectively sequestering interfering ions in high ionic strength environments. This integrated approach establishes a foundation for precision-engineered pollutant removal strategies in environmental remediation applications.
4.2
Heavy metal removal
The treatment of water contaminated with highly toxic heavy metal ions is critical to protecting biological systems and enhance environmental safety. While conventional DC electrochemical deposition, a common remediation method, unfortunately leads to energy waste and localized pH increase that block further deposition [110,111]. To address these issues, pulsed electrochemistry has been applied as a promising alternative, offering new insights into the elimination and capture of heavy metal ions by controlling the local environment [112]. Guo et al. developed a pulsed electrochemistry system featuring a periodically alternating applied voltage to selectively mitigate and capture heavy metals from complicated water systems (Fig. 3E) [108]. This system exhibited a greater removal efficiency for heavy metal (100% for Pb2+ and Cd2+, >98% for Mn2+) than other alkali and alkaline earth metal ions in a multicomponent solution. Results showed that the periodically alternating voltage notably suppressed water dissociation and overcame the mass transfer limitation of heavy metal ions diffusing to the electrode. However, challenges persist in large-scale applications due to electrode surface contamination, such as salt deposition and bubble formation, which can negatively affect the performance and long-term stability of electrode. The issues can be mitigated by using self-cleaning or anti-fouling materials like doped conductive polymers (e.g., polypyrrole, polyacetylene) [113-115]. As demonstrated by Yang et al., the incorporation of polypyrrole on electrode evidently improved the Faradaic pseudocapacitance, achieving 95.89% Cr6+ removal efficiency and maintaining stability over 200 cyclic voltammetry tests. This robust approach facilitated efficient Cr6+ removal in both aqueous and organic matrices, showcasing the versatility and reliability of polypyrrole-integrated electrodes for heavy metal remediation [116]. To facilitate large-scale implementation, the design of modular systems is essential. A feasible approach is to divide the system into independent functional modules, each of which enable ease maintenance and regeneration, minimizing the downtime of the entire system.
In addition, the pollution of crop soil with heavy metals is also a critical issue for the environment. The state-of-the-art electrochemical pollution treatment system employs asymmetrical pulse current to induce electroosmotic migration of heavy metal species to the negative electrode. At different initial concentrations, a variety of heavy metal species, including cadmium, lead, and copper, can be removed to a high degree in 0.5–6 h, meeting the regulatory standards for urban residential areas (Fig. 3F). Importantly, there was no secondary production of toxins and no excessive depletion of nutrients in the treated soil. A set of long-term studies and assays of plant life exhibit the impressive sustainability of the method and viability for agrarian applications [109]. Despite great progress, it remains challenging to achieve system simplification during the removal and recovery process of heavy metal. Particularly, integrating modular components into system design can facilitate easier maintenance for pulsed electrochemical system.
4.3
Nitrate reduction reaction
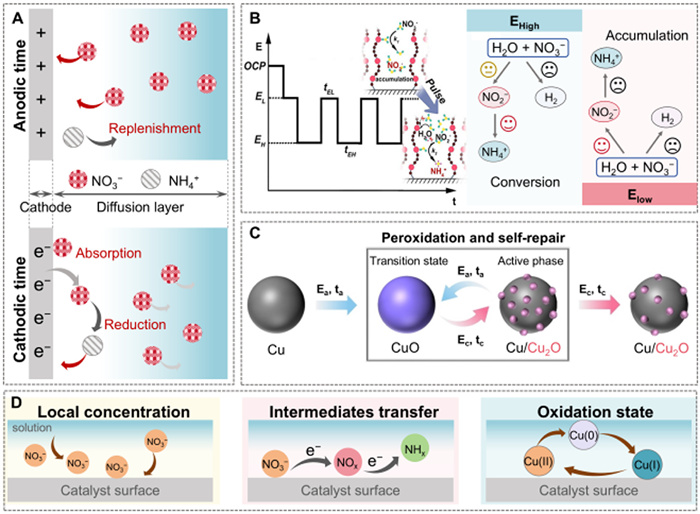
The elevated concentration of excessive NO3- ions in the global ecosystem disrupts the natural nitrogen cycle, posing significant ecological and human health risks. Hence, the development of reliable electrochemical methods with strong reactivity is of critical importance for the industrial use of NO3-. The efficacy of the pulsed electrochemistry strategy has well-documented to improve reaction selectivity, especially in cases that are hampered by the massive accumulation of NO3- near the electrode and competition from the hydrogen evolution reaction [97-99]. Huang et al. investigated the NO3RR kinetics by constructing a pulse-driven interfacial ion diffusion process (Fig. 4A) [100]. In situ Raman spectroscopy revealed that periodic anodic potentials replenished NO3- at the electrode surface, as indicated by the reappearance of the nitrate Raman peak (1050 cm−1) during the anodic phase. Conversely, cathodic potentials were responsible for depleting interfacial NO3-. Finite element analysis simulations further confirmed that pulsed operation maintained higher local NO3- concentrations in the diffusion layer compared to static conditions, circumventing mass transfer limitations and enhancing catalytic performance. Another typical pulsed electrocatalytic approach involves cascading the accumulation and conversion of NO2- intermediates during the NO3RR while blocking competition from the hydrogen evolution reaction, thus significantly improving the Faradaic efficiency (FE) and the ammonia (NH3) relative to DC electrolysis (Fig. 4B) [117]. Additionally, the surface reconstruction of catalysts driven by an electrical pulse can also promote the conversion of NO3- to NH3. An operando study demonstrated that pulsed conditions enabled pure Cu catalysts to (re)generate the desired oxidation state and surface structure [101]. The applied anodic potentials created a CuO sink, leading to abundant Cu/Cu2O complexes during reduction under a negative potential. This promoted a high yield and FE of NH3 formation in the optimized pulsed electrochemistry conditions (Fig. 4C).
Figure 4
Figure 4.
(A) Schematic illustration for the effect of pulse voltage on the mass transport of different species. Adapted with permission [100]. Copyright 2023, Springer Nature. (B) Reaction pathways of the NO3RR through the tandem NO2- accumulation-conversion process with pulsed electrolysis. Adapted with permission [117]. Copyright 2023, American Chemical Society. (C) In situ electrochemical peroxidation and self-repair of Cu under pulsed NO3RR at pH 14. Adapted with permission [101]. Copyright 2023, Wiley. (D) The main contributions of pulsed electrochemistry in NO3RR.
As discussed, pulsed electrochemistry contributes to NO3RR in three primary mechanisms: (ⅰ) Enhancing local *NO3- concentration, (ⅱ) accelerating intermediates transfer, and (ⅲ) creating a dynamic materials surface (Fig. 4D). Notably, highly reactive and unstable intermediates can rapidly decompose or transform, diminishing the overall efficiency. Additionally, unclear interfacial reaction kinetics also present significant challenges in pulsed electrocatalytic NO3RR. Stabilizing agents or surface modifications can help stabilize intermediates and enhance reaction efficiency. Furthermore, employing computational modeling enables researchers to predict reaction pathways and intermediates behavior, facilitating simulation of various conditions to identify optimal strategies. These combined approaches enhance overall efficiency and reliability in NO3RR under electric pulse action, paving the way for more effective environmental applications.
4.4
Carbon dioxide reduction reaction
Similar to the NO3RR, the activity of desirable products in the CO2RR can also be modulated through electrical pulse action. Cu-based catalysts have garnered significant attention as the only materials capable of efficiently producing C2+ products due to their optimal *CO binding energy. However, alternative metal catalysts have demonstrated superior performance for specific product profiles under pulsed operation. For example, Au exhibits exceptional selectivity toward formic acid [118], while Ag catalysts can also achieve high FE (up to 80%) for CO [119]. Conversely, Ni, Ti, Pt, and Fe catalysts remain impractical due to predominant hydrogen evolution behavior. Across the broad field of pulsed CO2RR, long-term stability persists as a critical bottleneck for sustainable CO2RR implementation. A pragmatic electrocatalyst must not only exhibit high product-specific activity and sustained selectivity under dynamic conditions, but also uphold such levels over extensive periods [120].
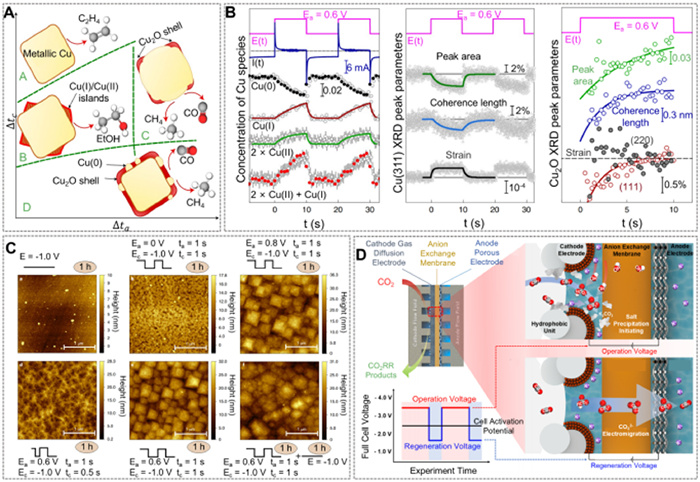
Currently, pulse-driven strategies in CO2RR primarily focus on the evolution of oxidation states, changes in catalyst surface morphology, and the rearrangement of ions in bulk solution [102,103]. In situ characterization by Timoshenko et al. proposed a relationship between the type and quantity of Cu oxide formed and the product selectivity under pulsed conditions (Figs. 5A and B) [121]. A larger cathode pulse times resulted in the FE of CO2RR products resemble to that of DC process, while shorter pulse times significantly affect the oxide formation on the catalyst surface. Specifically, the improvements in ethanol production with short anode and intermediate cathode pulsed durations were specifically achieved through a careful balancing of reduced and oxidized Cu species on the surface of the catalyst. Additionally, the existence of a distorted copper oxide phase also contributes to these enhancements. In a separate study using a single crystal of Cu(100), pulsed electrolysis was used to regularly regenerate morphological motifs and Cu(Ⅰ) species, leading to greatly increased selectivity for ethanol production when highly defective Cu(Ⅰ)/Cu(0) interfaces were available (Fig. 5C) [122]. Extensive researches have been dedicated to enhancing the selectivity for C2+ products by optimizing the arrangement of ions on the catalyst surface using pulse voltage. Xu et al. reported a pulsed self-cleaning CO2RR system that modified the arrangement of HCO3- and CO32-, further preventing saturation and avoiding the formation of CO32- (Fig. 5D). This study demonstrated stable C2+ products selectivity and partial current density, providing operation of the system with long-term stability [123].
Figure 5
Figure 5.
(A) Schematic depiction of the catalyst structure and composition during a cathodic pulse extracted. Δta: the duration of anodic pulse, and Δtc: the duration of cathodic pulse. (B) Variations in catalyst structure and composition during a voltage pulse. Reprinted with permission [121]. Copyright 2022, Springer Nature. (C) Electrode after electrolysis under different pulsed conditions. Adapted with permission [122]. Copyright 2020, Springer Nature. (D) Carbonate formation in membrane electrode assembly CO2 electrolyzes and the self-cleaning CO2 reduction strategy. Adapted with permission [123]. Copyright 2021, American Chemical Society.
Although efficient current use and low energy consumption have been accomplished, the regeneration of the electrodes is still a significant problem in pulsed CO2RR. Past researches have demonstrated that catalysts can experience alterations including surface reconstruction and aggregation during electrolysis, which can cause a rapid drop in electrode durability [69]. Meanwhile, precipitation of salts and flooding further decreased the longevity of gas diffusion electrodes at high current densities. The potential to enhance catalyst durability under pulsed condition has been seen in a series of regeneration experimental investigations. For example, the catalyst’s lifetime can be prolonged by combining short electrolysis periods with progressive chemical oxidation on the catalyst surface during regeneration segments [124]. During electrode regeneration, catalyst surface reconstruction driven by the electric field change under pulsed conditions promotes the migration and rearrangement of surface atoms, thereby changing the distribution of active sites on the catalyst surface. Despite substantially improvements of catalyst lifetimes, this periodic oxidative method can only slow, but not completely prevent, the electrode deactivation. Furthermore, Kok et al. found that migration of spatial Cu is slowed by oxidative pulses alone, potentially extending the ethylene yield (FE = 38%) versus the continuous case [104]. However, the prolonged regeneration cycles inevitably lead to catalyst performance decay due to electrode degradation (e.g., bond cleavage from repetitive redox cycles) and surface fouling (e.g., irregular re-deposition of reduced metal species or electrolyte-induced corrosion) [125,126].
Emerging strategies focus on integrating pulsed operation with surface engineering. To date, numerous reports have proven that rare-earth elements doping can improve the catalyst stability by modulating electronic structures to resist oxidative and corrosive damage [127]. Parallel efforts have also explored the anti-fouling coatings capable of repelling impurity adsorption or in situ decomposition of organic deposits under pulsed conditions [128]. These modifications synergize with pulsed regimes to preserve active site distribution and mitigate structural deterioration, offering pathways toward durable electrode design for industrial-scale CO2RR applications.
5.
Challenges and future prospects
Standing at the forefront of modern current regulation strategies, pulsed electrochemistry technology has emerged as a rising star, offering unparalleled efficiency, selectivity, and sustainability for water treatment. This review highlights the recent advances in fundamental principles, operation parameters, and practical application of the pulsed electrochemistry technology. Key parameters for optimizing pulsed electrochemistry performance are discussed. Additionally, potential environmental applications are also explored, including the degradation of persistent pollutants, recovery of heavy metals, regeneration of the electrode, and the electrocatalytic reduction reactions.
Although the exploration of pulsed electrochemistry has witnessed substantial advancements in electrocatalysis and shows promise for environmental applications, in general, its potential extends far beyond. Integrating pulsed electrochemistry with other advanced treatments presents a cost-effective and forward-looking approach for water treatment at a variety of scales. For example, combining it with membrane filtration can not only mitigate membrane fouling but also improve water flux through periodic electrical polarization. Similarly, its synergy with advanced oxidation processes establishes an energy-smart solution toward recalcitrant pollutant degradation, with precisely controlled electrical pulses optimizing radical yields. The integration with ozone-based systems enables spatiotemporal control of ozone utilization through pulse-modulated bubble generation and hydroxyl radical production. Coupling with persulfate activation processes may achieve on-demand sulfate radical generation via pulsed electron transfer, potentially overcoming current limitations in oxidant utilization efficiency. These advanced hybrid configurations hold the potential to establish adaptive treatment systems that can respond in real-time to fluctuating water quality through artificial intelligence-driven pulse parameter optimization. However, the complexity of designing a hybrid system demands meticulous optimization of operational parameters and a thorough understanding of synergistic effects for successful implementation. Achieving long-term operational stability will require the development of innovative electrode materials resistant to pulse-induced corrosion. Through fundamental mechanistic studies and cross-disciplinary collaboration will ultimately determine the commercial feasibility and environmental impact of next-generation pulsed electrochemical water treatment systems.
In advancing research in pulsed electrochemistry, artificial intelligence (AI)-driven optimization holds significant promise for enhancing treatment processes and allowing for real-time adjustments and more energy-efficient operations. Machine learning can process existing data to predict optimal operational parameters, paving the way for adaptive and self-optimizing water treatment systems. Additionally, AI can provide insight into pulsed electrochemistry reaction mechanisms at the atomic and molecular levels, refining models for large-scale applications. However, integrating machine learning with pulsed electrochemistry requires substantial computational resources and advanced algorithm development, necessitating collaboration among chemists, engineers, data scientists, and environmental scientists to achieve these objectives.
Scaling up of pulsed electrochemistry from laboratory research to industrial water treatment demands coordinated advancements in technical, economic, and operational aspects. A few commercial systems, such as the advanced pulsed electrochemical technology for textile wastewater treatment, have been developed to address complex effluents containing recalcitrant pollutants. However, independent verification on efficiency (e.g., energy consumption, removal rates) and cost-effectiveness is currently lacking. Transitioning this technology to practical implementation involves overcoming three core challenges: (ⅰ) Economic viability requiring cost-benefit analysis of pulsed generator, energy consumption, and electrode maintenance versus conventional methods; (ⅱ) Scalability through modular reactor designs and distributed units to handle variable throughput; (ⅲ) Operational stability ensuring durable electrodes and adaptive pulse controls for fluctuating water quality. Standardization efforts are crucial to unify protocols for parameter optimization, monitoring, and effluent validation. Interdisciplinary collaboration will be essential to translate academic innovations into practical solutions that balance environmental benefits and economic feasibility for large-scale implementation. This collaborative effort will be pivotal in advancing pulsed electrochemistry wastewater management on a global scale.
In conclusion, this work offers a comprehensive overview of the fundamental understandings, operational parameters, and environmental applications of pulsed electrochemistry strategy for water treatment, highlighting new insights and challenges. Instead of pointing to one single mechanism, in this review, we present a broad framework for the use of pulse as important variables to optimize electrocatalytic performance. Hopefully this information will help transfer pulsed electrochemistry systems from experimental innovations to practical, cost-effective for environmental solutions.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by the by the National Natural Science Foundation of China (No. W2412093) and the Natural Science Foundation of Shanghai (No. 23ZR1401300).
Figure 2
(A) The typical waveforms of pulsed electrochemistry. (B) Applied potential and current response curves under different pulse waveforms. (C) Hydrogen production rate versus pulse frequency under different pulse waveforms. Reprinted with permission [38]. Copyright 2024, Elsevier. (D) Unidirectional and (E) bidirectional square wave pulse with periodic switching potential. Adapted with permission [84]. Copyright 2023, Elsevier. (F) Schematic diagram of in situ pulsed synthesis of tungsten carbide. Adapted with permission [82]. Copyright 2022, Elsevier. (G) Schematic diagram of the pulsed electrochemical oxidation process for biologically treated leachate treatment. Adapted with permission [83]. Copyright 2021, American Chemical Society.
Figure 3
(A) Time course of phenol oxidation during direct-current and pulsed-current electrolysis as a function of current density. Adapted with permission [94]. Copyright 2021, American Chemical Society. (B) Acid Blue 9 and COD removals at pulse and DC modes (The inset panel gives the corresponding the electrical efficiency per log order reduction value at pulse and DC modes). Adapted with permission [87]. Copyright 2021, Elsevier. (C) Comparison of performance of different oxidation processes were conducted regarding removal of sulfamethoxazole. Adapted with permission [95]. Copyright 2023, Elsevier. (D) The schematic illustration of pulse-on time and pulse-off time during pollutants degradation. Adapted with permission [94]. Copyright 2021, American Chemical Society. (E) Conversion process of heavy metal ions during pulsed electrodeposition. Adapted with permission [108]. Copyright 2024, American Chemical Society. (F) Working principle of the pulsed remediation method for heavy metal removal. Adapted with permission [109]. Copyright 2019, Springer Nature.
Figure 4
(A) Schematic illustration for the effect of pulse voltage on the mass transport of different species. Adapted with permission [100]. Copyright 2023, Springer Nature. (B) Reaction pathways of the NO3RR through the tandem NO2- accumulation-conversion process with pulsed electrolysis. Adapted with permission [117]. Copyright 2023, American Chemical Society. (C) In situ electrochemical peroxidation and self-repair of Cu under pulsed NO3RR at pH 14. Adapted with permission [101]. Copyright 2023, Wiley. (D) The main contributions of pulsed electrochemistry in NO3RR.
Figure 5
(A) Schematic depiction of the catalyst structure and composition during a cathodic pulse extracted. Δta: the duration of anodic pulse, and Δtc: the duration of cathodic pulse. (B) Variations in catalyst structure and composition during a voltage pulse. Reprinted with permission [121]. Copyright 2022, Springer Nature. (C) Electrode after electrolysis under different pulsed conditions. Adapted with permission [122]. Copyright 2020, Springer Nature. (D) Carbonate formation in membrane electrode assembly CO2 electrolyzes and the self-cleaning CO2 reduction strategy. Adapted with permission [123]. Copyright 2021, American Chemical Society.
(ⅰ) Achieving the in situ stabilization of oxygen-containing functional groups active sites; (ⅱ) Updating the interface microenvironment to promote O2 absorption and proton transfer
A 210% leap in H2O2 yield and a 74% increase in FE

 DownLoad:
DownLoad:

 下载:
下载:






 下载:
下载:
