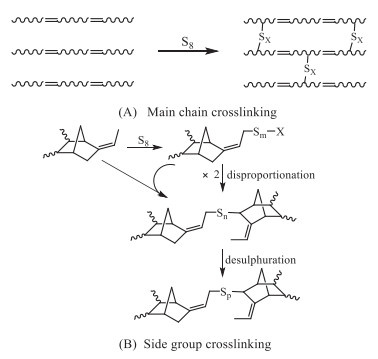
Scheme 1.
Sulfur-vulcanization of the unsaturated carbon-chain rubbers.

As the most principal kind of rubbers, carbon-chain rubbers have been widely used in many fields including transportation, medicine, construction, and everyday life, owing to their excellent flexibility and low-cost. They could be classified into natural rubbers and synthetic rubbers according to their sources, while the synthetic rubbers cover the unsaturated and saturated ones based on their carbon chains.
To achieve the desired flexibility and mechanical property, the rubber molecules are usually crosslinked as vulcanized rubbers, to restrict the slide of the rubber molecules from each other. However, the chemical crosslinking makes the vulcanized rubbers insoluble and non-melting. As a result, the end-of-life products or production scrap could not be recovered and reused by facile re-processing, like the linear and branched polymers. By now, landfill and incineration are the most widely used methods to treat the waste rubbers all over the world [1], causing serious environmental pollution and resource waste.
In the last decades, the waste vulcanized rubbers have been widely used in the construction and road materials [2], as well as the fillers in polymer materials, including polymer-based composites [3]. Although such solid wastes could be temporarily re-used by these methods, the risk of environmental pollution remains. Alternatively, the waste vulcanized rubbers could be thoroughly eliminated with microbial degradation [4], however, leading to resource waste.
Besides, high-value recovery of waste rubbers could be achieved by the chemical recovery. By now, there were several excellent reviews on the treatments of the waste vulcanized rubbers, mainly on the treating methods by various physical and chemical processes [5-10]. Different from the hydrocarbons for energy recovery [11,12] and carbon source for the synthesis of nano-carbons [13,14] via pyrolysis, the waste vulcanized rubbers could be devulcanized and degraded into soluble polymers and oligomers by the mechanochemical [15] and thermochemical [16] approaches, which could be used in the synthesis of new polymers. It has been recognized as the most promising strategy in the treatment of the waste vulcanized rubbers, as a versatile resource recycling. However, the random scission of chemical bonds in these methods makes the molecular structures of the degraded products more complicated, although the bond energies of the crosslinking bonds such as C-S (285 kJ/mol), S-S (268 kJ/mol) and -Sx- (251 kJ/mol) were lower than the carbon chain bond of C—C (346 kJ/mol), the number of C—C bonds is much higher than the others. Moreover, the undesired re-crosslinking would occur in the radical recombination [8].
For re-vulcanized rubbers with better mechanical and flexible performance, the chemically degraded products are desired with definite molecular structures, for a better application as structural prepolymers in the synthesis of new polymer products. In the present work, the recent progress in the chemical recovery of the waste sulfur-vulcanized carbon-chain rubbers was reviewed, emphasizing the molecular structures of the chemically devulcanized products for reuse. The restrictions and challenges in the application of these degraded products were also proposed, as well as the future perspectives.
The traditional vulcanized carbon-chain rubbers are usually produced by the vulcanization of the linear carbon-chain rubbers with different crosslinkers, except the self-crosslinking ones [17]. The saturated linear carbon-chain rubbers without C=C group are usually vulcanized with organic peroxides via the chain transfer of the primary free radicals and then coupling termination. The resultant crosslinked framework is completely formed with C—C bond, which could hardly be selectively broken. So, the sulfur-free vulcanized saturated carbon-chain rubbers could only be recovered as random oligomers, along with undesired re-crosslinking. The inhomogeneous crosslinked structure would be therefore resulted in the re-vulcanized rubbers, declining their flexibility.
For the unsaturated rubbers containing C=C groups in their main chains (natural rubber and synthetic diene-based rubbers including butyl rubber) or side groups (ethylene propylene-diene (EPDM)-rubber), sulfur is the most widely used crosslinker. According to the site of the C=C groups, they could be classified as main chain crosslinking type and side group crosslinking type for the ones with C=C groups in their main chains (natural rubber, synthetic diene-based rubbers and butyl rubber) or side groups (EPDM), respectively (Scheme 1). After the vulcanization, the polysulfide crosslinking chains are introduced into the products. Therefore, there are polysulfide crosslinking chains and carbon chains in the sulfur-vulcanized carbon-chain rubbers, interconnected with each other via chemical bond. To devulcanize these vulcanized rubbers, both the polysulfide crosslinking chains and carbon-based main chains might be cleaved, denoted as crosslink breakage (devulcanization) and main-chain scission (degradation) according to the Horikx's analysis [18].
The chemical recovery of the waste sulfur-vulcanized carbon-chain rubbers is denoted as chemical treating of the waste sulfur-vulcanized carbon-chain rubbers into soluble polymers and/or oligomers in molecular level, which could be used as prepolymer for new rubbers via facile re-vulcanization. Therefore, the chemical devulcanization and degradation should be occurred on the unique chemical bonds via selective scission, distinctly different from the random scission in the energy-dependent reclamation method, including heating, shearing and grinding.
The radical reactions have been widely used for the devulcanization of the sulfur-vulcanized carbon-chain rubbers, by selective scission of the C-S bond or S-S bond in the crosslinking chains. By the means, linear or slightly branched polymers were obtained, which could be re-vulcanized as new rubbers.
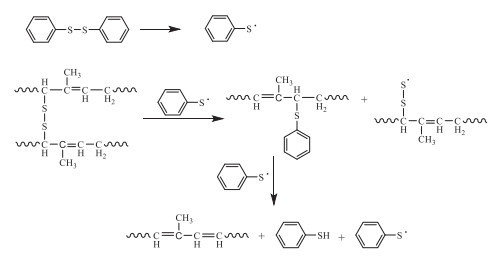
Organic disulfides, especially diphenyl disulfide (DD), have been used for the devulcanization of the sulfur-vulcanized carbon-chain rubbers by selective scission of the C-S bond in the crosslinking chains (Scheme 2).
Jana et al. reported the mechano-chemical recycling of sulfur-vulcanized natural rubber with or without DD at 120 ℃ for 10 min on a two-roll cracker-cum mixing mill. The obtained devulcanized product was re-vulcanized again by the addition of sulfur and N-cyclohexyl-2-benzothiazyl sulfonamide (CBS). By the approach, the mechanical properties of the sulfur-vulcanized natural rubber were retained in more than 87%, including 100% modulus, 200% modulus, tensile strength, elongation at break and tear strength [19]. It was found that the mechanical properties including moduli, tensile strength, tear strength, elongation at break were higher for the revulcanized rubber obtained in the presence of DD, than that obtained in absence of DD, due to the higher crosslinking density in the former one. The authors claimed that the extra crosslinking in the revulcanized rubber was caused by the formation of new active crosslinking sites via chain transfer reaction in the devulcanization.
It should be noted that the byproducts such as hydrogen sulfide and thiols were released in the chemical devulcanization with DD [20]. The disulfide and polysulfide crosslinking chains could be selectively cleaved with thiols in presence of organic bases via nucleophilic displacement reaction, while the monosulfide crosslinking chains were more stable and not affected. Furthermore, the carbon chains would be broken off by increasing the devulcanization temperature, while declining the mechanical performance of the resultant products [5].
Kojima's group reported the devulcanization of different rubbers in an autoclave with supercritical carbon dioxide (scCO2). It was found that the sol component yield increased with increasing the CO2 pressure for the unfilled vulcanized polyisoprene (PI) rubber [21] and natural rubber (NR) with DD as decrosslinking reagent [22]. The selective impregnation of decrosslinking reagent (DD [23] or dicumyl peroxide (DCP) without sulfur to simplify the analysis [24]) into the vulcanized rubbers via the supercritical CO2 was also revealed.
These works revealed the devulcanization of the disulfide and polysulfide crosslinking chains, but not the monosulfide crosslinking ones. Moreover, the devulcanized products showed a higher glass transition temperature (Tg) than the virgin rubber, due to the pendant phenyl side groups. In the devulcanization of waste tread rubber in supercritical carbon dioxide [25], the cyclic sulfide structures were revealed as another reason for the increased Tg.
It has also been reported that the presence of fillers such as carbon black and silica in the vulcanized NR or synthetic rubbers did not affect the devulcanization with DD, and the tensile properties of recycled rubber vulcanizates made from virgin rubber and 20–60 phr of recycled rubber were over 90% of those of the original vulcanizates [26,27].
Mohaved et al. reported the devulcanization of discarded EPDM automotive parts with 2-mercaptobenzothiazole (MBT) or tetramethylthiuram disulfide (TMTD) as devulcanizing agent, respectively [28]. MBT was found to be more efficient than TMTD. The results indicated that there was no adverse effect on the scorch and optimum cure times, crosslink density, rate of cure, and viscosity by replacing 60 wt% of the virgin rubber in the automotive rubber strips with the devulcanized powder. Moreover, the hardness, compression set, and modulus at 20% elongation were unaffected for the re-vulcanized product when 20 wt% of the virgin rubber was replaced.
Ghorai et al. reported the mechanochemical devulcanization of sulfur-vulcanized NR in an open roll mixing mill with bis(3-triethoxysilyl propyl)tetrasulfide (TESPT) as dual functional devulcanizing agent, both the disulfide-based devulcanizing agent and coupling agent for dispersion of silica [29]. Due to the mechanical milling, the tetrasulfide bond in TESPT and the C—C and C-S bonds in the sulfur-vulcanized NR could be broken, therefore, the functionalized linear and branched devulcanized products were obtained via the coupling of the radicals formed (Scheme 3). Prolonging the milling time from 20 min to 60 min, the sol content increased from 34.8% to 42.8%, and the Tg increased from 207.7 K to 213.9 K, due to more silane groups had been introduced, which was revealed by the increased S content in the devulcanized products increasing from 2.98% to 3.21%.
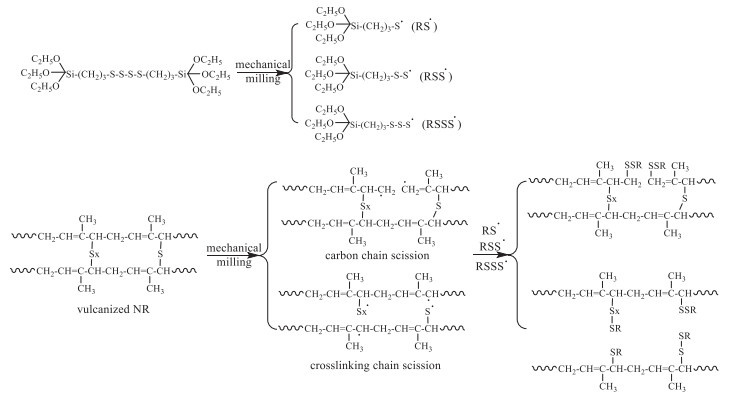
The resultant functionalized linear and branched devulcanized products were re-vulcanized with or without silica as filler. It was found that the optimum cure time decreased whereas cure rate increased with prolonging the devulcanization time. The mechanical properties, including modulus at 50% and 100% elongation, tensile strength and elongation at break, increased with devulcanization time up to 40 min with 6 mL TESPT/100 g ground NR.
Organic peroxides have been reported for the devulcanization of the sulfur-vulcanized carbon-chain rubbers by selective scission of both weak bonds (S-S and C-S) in the crosslinking chains (Scheme 4).
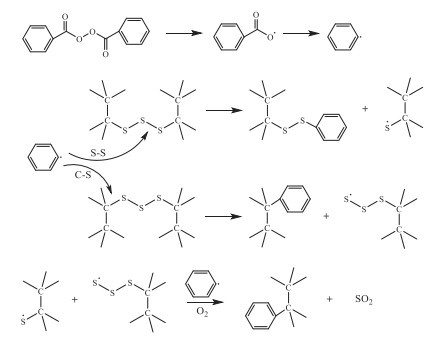
Rooj et al. reported the devulcanization of NR with benzoyl peroxide (BPO) as devulcanizing agent [30]. Prolonging the treating time, the absorbance intensity of S-S at 556 cm-1 declined in the ATR-IR analysis, while the characteristic absorbance peaks of C=C at 837 cm-1 and -CH3 at 1376 cm-1 remained constant, revealing the devulcanization by the S-S scission. Moreover, the weight average molecular weight (Mw) of the devulcanized products was measured as 3.3 × 105 g/mol by the viscosity method after devulcanization at 80 ℃ for 6 h, very near to the virgin NR, meaning the carbon chains were maintained in the devulcanization.
The devulcanized natural rubber (DNR) was then co-vulcanized with the virgin natural rubber with different proportions, 20DNR/80NR, 40DNR/60NR, 60DNR/40NR and 80DNR/20NR, respectively. The highest tensile strength and modulus at 300% elongation were obtained with the 40DNR/60NR, much higher than the original NR sample. It might be resulted from the introducing of phenyl groups in the DNR. While the data decreased sharply with further increasing the DNR proportion, it was explained with the poor dispersion of DNR in the rubber matrix, which would cause numerous stress concentration points in the re-vulcanized DNR/NR.
In a following work, it was found that the selective S-S scission could be achieved at devulcanization time of 2 h with lower BPO concentration, while the non-selective chain scission including the carbon chains occurred with higher temperature, longer time and higher BPO concentration [31]. The re-vulcanized samples showed the lower tensile strength and percentage of elongation at break in comparison with the original one. A proportion of 40 wt% was also recommended for the blend with virgin rubber, without adverse effect on the mechanical properties of the re-vulcanized rubber.
Dez's group reported the recovery of functionalized rubber from waste tires by radical devulcanization with different peroxide derivatives as the devulcanizing agent [32], such as halogenated aromatic peroxides, ester-functionalized peroxides, alkyl aromatic peroxides and nitro-functionalized peroxides, respectively. As a result, the halogenated polymers, or polymeric acids, alcohols or amines could be obtained, for applications in supramolecular interactions, surfactants, or polyelectrolytes.
Colom et al. investigated the devulcanization process of scraps of EPDM systems from commercial, industrial and residential roofing systems by combined thermomechanical/microwave (TM/MW) procedure with BPO as devulcanizing agent [33]. It was found that the combined approach acted more selectively on the disulfide bonds and sparing the main polymeric chain, compared with the TM one, according to the Horikx's theory. As a result, a product that was more like the original fresh EPDM could be obtained with 2 phr of BPO. As for the ground tire rubber (GTR), the combined TM/MW devulcanization process resulted in greater degradation of the main rubber chains in the cross-linked network compared to the process using only MW [34].
Dukhuis et al. reported the reclaiming of sulfur-vulcanized EPDM rubbers with hexadecylamine (HDA) [35]. It was found that the devulcanization efficiency was determined by the crosslink density and crosslink distribution. And temperature was found as the main governing factor and addition of HDA could significantly enhance the effects. Different from the thermal treatment without HDA via the random carbon-chain scission of the rubber network, reclaiming of the conventionally vulcanized compound was mainly occurred at the poly- and disulfidic crosslinking chains, by breaking and transforming into monosulfidic bonds, while that of the efficiently vulcanized compound primarily happened at the carbon chains, because it was mainly crosslinked with the monosulfidic bond with higher dissociation energy than the di- and polysulfidic ones.
For both sulfur-vulcanized EPDM rubbers, the proportion of the devulcanized products up to 50 wt% led to a mild influence on the mechanical properties of the re-vulcanized rubbers. As for the ground non-reclaimed rubber powder, more than 10–15 wt% in the virgin compounds led to significant property decline. The results indicated that the side group crosslinking type rubbers could be also chemically recovered, despite of the various devulcanization mechanisms for the waste rubbers with different crosslinking structures.
In the engineering kinetic modeling on the EPDM devulcanization in an internal batch mixer with HDA as the devulcanizing agent [36], the rate of side reactions involving HDA was much lower than the reaction rate between HDA and cross-links at the HDA concentration studied, by comparing the calculated activation energies of the devulcanization reactions on the polysulfidic, disulfidic and monosulfidic cross-link scission and main-chain scission.
Movahed et al. reported the devulcanization of waste EPDM by microwave radiation with different devulcanizing reagents, such as HDA, N-cyclohexyl-2-benzothiozylsulfenamide (CBS), dipentamethylenethiuram tetrasulfide (DPTT), MBT, 2-mercaptobenzothiazole disulfide (MBTS), and TMTD [37]. The highest devulcanization was obtained with HDA at about 91%-94% with increasing temperature from 200 ℃ to 260 ℃. A nucleophilic devulcanization mechanism had been suggested with the α-H-containing aliphatic amines such as HAD, while a radical mechanism was proposed for devulcanization with other five devulcanizing reagents. Although the mechanical properties of the re-vulcanized rubber compounds were lower than the virgin EPDM, they could be used for the blends with the virgin EPDM. The mechanical properties including hardness, tensile strength, compression set, and elongation at break of 75:25 virgin EPDM/devulcanized rubber blend were comparable with those of the vulcanized virgin EPDM. However, the hardness and compression set of the 50:50 blend were slightly inferior compared with those of the vulcanized virgin EPDM. The differences in the mechanical properties were explained with cross-linking density of the re-vulcanized rubbers.
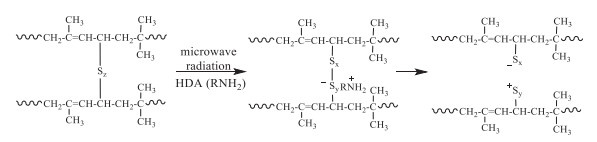
Khavarnia and Movahed reported the devulcanization of butyl rubber (IIR) by microwave radiation with different devulcanizing reagents. Compared with DD, CBS, and TMTD, HDA exhibited the highest devulcanization of 83%, with 30 and 6 phr of paraffinic oil and HDA at 180 ℃ [38]. It was found that the disulfides crosslinking bonds had been partially broken and new mono and polysulfidic bridges were formed by releasing sulfur (Scheme 5).
In the comparative investigation on the reclamation of NR based latex products with HDA and DD [39], it was found that reclamation has mainly occurred through the crosslinking chain scission rather than main-chain scission with DD by breaking all poly- and disulfidic crosslink chains, while the formation of additional crosslinking chains was found with HDA, as some of the polysulfidic crosslinks remained in the reclaimed product.
It has also been reported that the scCO2 could effectively improve the diffusion of devulcanizing reagents into unfilled polyisoprene rubber (IR) vulcanizates [21]. And the thiol−amine reagent (PhSH/n-BuNH2) was found to be effective among several devulcanizing reagents, especially for the IR vulcanizates with shorter cure time or higher sulfur/MBT.
Besides the small molecular amines, the polyurethane aminolysis products (PAPs) was also be used as amine-type devulcanizing agent for the devulcanization of waste tires [40]. The addition of PAPs could shorten the optimum cure time of the reclaimed rubbers (RRs) and improve the crosslink density and hardness of the RR re-vulcanizates. Most recently, the group reported the devulcanization of the scrap truck tire in low-temperature mechano–chemical process at 60 ℃ with alcoholic amines as the devulcanizing agent, including hydroxyethyl ethylenediamine (AEEA), ethanolamine (ETA), and diethanol amine (DEA) [41]. The results indicated that the amino groups in the alcoholic amines could react with sulfur after the crosslinking bonds were broken by mechanical shear force, thus blocking the activity of sulfur and introducing hydroxyl groups into the rubber chains, enhancing the devulcanizing degree and devulcanizing efficiency, reducing the Mooney viscosity, and improving the mechanical and anti-aging performance. Compared with the product obtained without alcoholic amine, the sol content of reclaimed rubber increased from 13.1% to 22.4%, the devulcanization ratio increased from 82.1% to 89.0%, the Mooney viscosity decreased from 135.5 to 83.6, the tensile strength improved from 14.7 MPa to 16.3 MPa, and the retention rate of tensile strength raised from 55.2% to 82.6% after aging for 72 h, while the devulcanization time was shortened from 21 min to 9.5 min, with DEA as the devulcanizing agent.
The oxidation method has been widely reported with various inorganic oxidants. However, the oxidation, scission and recombination occurred simultaneously by the non-selective scission of the carbon chains and crosslinking chains, thus, the oxygen-containing groups such as hydroxyl, carbonyl, carboxyl, sulfoxide and nitro groups were resulted in the devulcanized products [42-45].
For example, Wang's group reported the thermal-oxidative reclamation of waste tire rubbers for producing high-performance rubber composites, by thermal oxidation of ground tire rubber at 200 ℃ [45]. High scission efficiency was obtained with a sol fraction of 66.53 wt% after 20 min without any chemical agent. The devulcanized product containing the soluble sol and ultrafine fillers could be easily dispersed into NR for producing composite. The mechanical properties and aging resistance of the composite were enhanced with the addition of up to 40 wt% of the reclaimed ground tire rubber, owing to the antiaging agent and carbon black in the waste tires, as well as a lower content of C=C in the devulcanized sol. However, increasing the proportion of the devulcanized product caused a sharp decline in the mechanical properties of the resultant composite, because of the poor dispersibility of the devulcanized product in NR, due to the oxygen-containing groups formed in the thermal-oxidative reclamation.
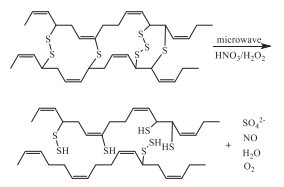
Valdes et al. reported the synergism between HNO3 and H2O2 on the devulcanization of sulfur-vulcanized rubber under microwave irradiation (Scheme 6) [46]. 5 mol/L H2O2/1% HNO3 was revealed with the better devulcanization efficiency. Increasing HNO3 concentration led to a carbon chain scission, while better devulcanization via the crosslinking chain scission was resulted by increasing the H2O2 concentration.
Buitrago‑Suescún and Britto reported the devulcanization of the waste ground tire rubber by combination of oxidation with potassium permanganate/hydrogen peroxide, followed by microwave exposure in the presence of MBT as devulcanizing agent [47]. The combination of both methods has been analyzed by using the devulcanized rubber as raw materials in proportions greater than 100 phr in new styrene-butadiene rubber (SBR) rubber compounds with tensile strength, elongation at break and tear resistance properties like the vulcanized SBR/silica composite.
With the assistance of soybean oil, H2O2-induced thermo-oxidative reclamation of vulcanized SBR at 100 ℃ [48] and thermo-oxidation of butadiene rubber (BR) vulcanizate with air at 140 ℃ [49] could be efficiently enhanced, by suppressing the recombination of rubber chains.
In the unsaturated rubbers containing plentiful C=C groups in their carbon chains, such as NR and the main synthetic diene-based rubbers including polyisoprene rubber, polybutadiene rubber, SBR and nitrile rubber (NBR), these C=C groups could be broken for the devulcanization. Although the C=C bond energy (611 kJ/mol) is much higher than the C—C bond, it possesses a higher activity due to the π bond. So, the waste sulfur-vulcanized unsaturated rubbers could be easily degraded into soluble sols by selective breaking the C=C groups in their carbon chains.
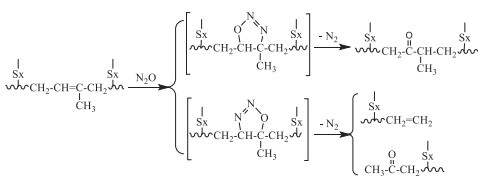
Dubkov and coworkers reclaimed the scrap tire rubbers by treating with nitrous oxide (N2O) at 2–5 MPa at 230 ℃ for 12 h, by selective scission of the carbon chains, via a 1,3-dipolar cycloaddition of N2O to the C=C groups of isoprene and butadiene units in vulcanized elastomers (Scheme 7) [21,50]. The ketone-containing oligomers were obtained in the sol fraction with number average relative molecular weight (Mn) of 1200–1300. Owing to the sufficient amount of residual C=C bonds, the oligomers were expected to be re-vulcanized and reused for the partial replacement of virgin rubber in rubber compounds.
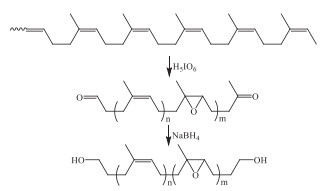
Oxidative scission of the C=C groups in the carbon chains of the diene-based rubbers have been widely used for the design of telechelic polymers with various osidants [51-54]. Pilard and coworkers reported the devulcanization of waste tires via the selective oxidative scission of the C=C groups with periodic acid. It was found that the C-S bonds were remained while the S-S bonds had been broken into -SO2R or -SO2OH groups, alone with the oxidative scission of the C=C groups [55]. Such side reaction due to the relatively lower oxidation selectivity caused the uncertainties in the molecular structure of the degraded products, which would also restrict their practical application. After reducing the carbonyl groups into hydroxyl groups (Scheme 8), the resultant hydroxyl oligomers were used as polyols in the preparation of polyurethane (PU) foams.
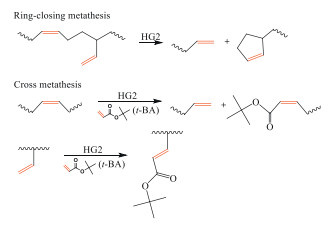
Metathesis reactions, involving a cyclic process where the C=C bonds are broken and restored, have been widely used in the functionalization of diene-based rubbers [56]. Wolf and Plenio reported the ethenolysis of end-of-life tire granulates with ethylene as chain transfer agent (CTA), catalyzed with ruthenium-based catalysts [57]. The yield was obtained around 50% as the soluble sols with degree of polymerization (DP) around 20. The C/H ratio of the degraded products was 5:7.8, very close to the theoretical value of 5:8 for polyisoprene, while the S content remained the same as in granulates. Such results indicated that only the C=C bonds in the mainchains has been broken via the ethenolysis, without any new terminal groups introduced in the products.
Recently, the technology has been successfully utilized in the degradation of the waste rubbers, especially the NR and cis-1,4-polyisoprene, owing to their plentiful cis-1,4-butadiene units in their carbon chains with different CTAs (Scheme 9) [58]. Owing to the new terminal groups had been introduced in the cross metathesis reaction, the degraded products could be used as functionalized liquid rubber [59], surfactant [60], corrosion inhibitor or adhesive [61], telechelic oligomers for innovative polymeric materials [62,63], and so on.
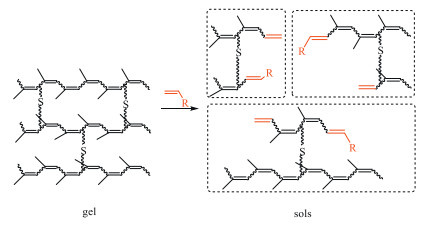
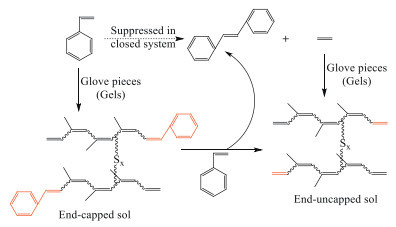
Besides the new terminal groups introduced in the cross metathesis, cyclic polymers would also be formed by the self-metathesis reaction between the resultant sols. Moreover, the self-metathesis reaction between CTAs would also restrict the metathetic devulcanization of waste rubbers [58]. To suppress self-metathesis reaction between CTAs, traceless metathetic degradation of waste natural rubber gloves with styrene in closed system was reported by our group [64]. Compared with the end-capped products with the styrene residues in the metathetic degradation with less styrene, the end-uncapped sols were obtained with excessive styrene through the two routes: i) ethenolysis with ethylene as CTA and ii) cross metathesis reaction between the styrene residues in the degraded products with the excessive styrene, eliminating the styrene residues (Scheme 10). Additionally, the potential application of the traceless-metathetic degraded product was proposed by facile crosslinking with an initiator for radical polymerization, 2,2′-azobis(2-methylpropionitrile) (AIBN). After heating at 70 ℃ with 2% AIBN, the degraded product lost fluidity and became an elastic solid, revealing the successful re-vulcanization.
Afterwards, the metathetic degradation was used for the recovery of the waste sulfur-vulcanized NBR [65]. As a synthetic diene-based rubber via radical emulsion polymerization, there is less cis-1,4-butadiene unit than the NR, the ring-closing metathesis involving the adjacent cis-1,4-butadiene unit and 1,2-butadiene unit was found (Scheme 11), favoring the metathetic degradation. This work is expected to open new idea for the recovery of the waste vulcanized emulsion-polymerized diene-based rubbers.
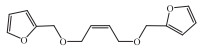
Another successful chemical recovery of the waste sulfur-vulcanized carbon-chain rubber was reported by the metathetic degradation of the vulcanized rubber with a furan allylic chain transfer agent (Fig. 1), and the furan functionalized polymers with Mn of less than 2 × 104 and a narrow polydispersity were used as secondary raw materials for elastomeric networks fabrication through the Diels−Alder (D-A) reaction with a tri-maleimide crosslinker [66]. The total tire waste-based network was synthesized with excellent reprocessing ability after 3 cycles.
Although the waste sulfur-vulcanized carbon-chain rubbers could be devulcanized and degraded by selective chain scission, there are still some challenges for their practical recovery.
The selective scission of the C-S and S-S bonds in the crosslinking chains would result in the linear polymers, while the branched polymers were obtained from the selective scission of the C=C bonds in the carbon chains (Scheme 9). Theoretically, the former ones are expected to be more promising for the chemical recovery of the waste rubbers for new rubbers with better mechanical and flexible performance, in comparison with the latter ones, which would lead to a non-uniform crosslinking in the re-vulcanized rubbers, although the metathetic approach in the latter ones is much milder.
Actually, the extra crosslinking in the revulcanized rubber was also found, due to the formation of new active crosslinking sites by the chain transfer reaction in the devulcanization [23]. Moreover, the active radicals were produced during the devulcanization with the organic disulfides and peroxides as the devulcanizing agent to break the C-S and S-S bonds in the crosslinking chains, which might cause the crosslinking of the devulcanized products to form the new crosslinked structure via C—C bonds, through the radical addition or chain transfer reactions. Although such structures had not been reported in the references, they are much difficult to be recovered than the ones crosslinked with the C-S and S-S bonds.
The cyclic sulfide structures and cyclic polymers would also be formed by the devulcanization with organic disulfide and the self-metathesis reaction between the resultant sols in the metathetic degradation. Furthermore, the pendant C-S-S-pH groups could also be formed in the devulcanized products of the crosslinked structure via mono-sulfide bond, according to the proposed mechanism (Scheme 2). Such side group might cause the additional crosslinking via disulfide metathesis reaction in the de-vulcanization, as a reverse reaction for the devulcanization.
The last challenge in the selective scissions is their selectivity. For example, the C-S bonds would also be broken during the oxidative scission of the C=C bonds [47], such uncertainties in the molecular structure of the degraded products would restrict their practical application.
Although the devulcanization via cleaving crosslinking linkers is usually accompanied by the main-chain degradation, linear rubbers like the original fresh ones could be obtained via the selective devulcanization of the waste sulfur-vulcanized carbon-chain rubbers [34], which could be easily re-vulcanized into new rubber products.
However, the branched rubbers are usually resulted via the main-chain degradation in the initial stage. The branched and/or cyclic liquid rubbers as prepolymers or oligomers would be caused by the main-chain degradation of the degraded products, even as small molecules in an extreme main-chain degradation [58,64,65]. Such small product could hardly be recovered in new rubber products, because they could hardly be separated and collected from the degraded mixture, causing the decline in yield.
Except the ethenolysis [57] and the traceless metathetic degradation [64], new terminal groups or side groups would be introduced in the devulcanized and degraded products. The polar oxygen-containing groups decline the dispersibility of the devulcanized and degraded products in the non-polar carbon-chain rubbers. And the introduced rigid groups, such as phenyl group, had been revealed to increase the Tg of the devulcanized products [25,29], declining the flexibility of the re-vulcanized rubbers.
The chemical recovery of the waste sulfur-vulcanized carbon-chain rubbers is mainly determined by the molecular composition and topology of the devulcanized and degraded products. Therefore, the future development in the topic should be focused on the selective chain scission with high selectivity, to ensure the molecular composition. On the basis, the new terminal groups or side groups should be averted, at least, the polar terminal groups or side groups should be avoided.
Facing the above challenges, a promising fundamental solution to the above problems should be the utilization of the carbon-chain rubbers without stable covalent crosslinking, such as the polyolefin thermoplastic elastomers by physical crosslinking via phase separation [67], or the chemical crosslinking via dynamic covalent bonds [68], such as disulfide bond [69], exchangeable β‑hydroxyl ester [70], reversible D-A adduct formation [71], metal-ligand interaction [72], and multiple dynamic interactions [73,74], for a self-healing and reprocessable characteristics.
Among them, the disulfide metathesis has been widely investigated in the self-healing and reprocessable rubbers, owing to its lower healing temperature than the DA reaction. Moreover, the repair of fracture surfaces or damage can be driven by heating, photolysis, or oxidation (Scheme 12).
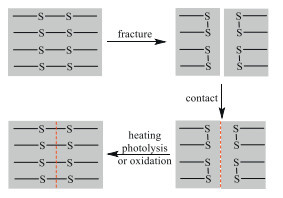
In the vulcanization of the carbon-chain rubbers with sulfur, the monodisulfide, disulfide and polysulfide bonds are formed as the crosslinking chains. For the recyclable sulfur-cured rubbers, Cu(Ⅱ) ion has been found as a catalyst to catalyze the disulfide metathesis reaction of inherent disulfide and polysulfide bonds [75], especially the organic complex copper(Ⅱ) methacrylate (MA-Cu) [76,77]. It was also found that the incorporated terminal hydroxyl and pyridyl groups in the cured rubber could reduce the catalytic activity of Cu2+ at service temperature by coordinate interactions, thus improving the creep resistance [78]. The resulting networks exhibited superior creep resistance even at a service temperature of 100 ℃, while the corresponding network rearrangement ability at the elevated temperatures was not affected. Moreover, the recycled samples displayed excellent mechanical properties with tensile strengths exceeding 7 MPa and the elongation at break over 600%.
It seems as promising choice by using the disulfide reversible chemistry in rubber to combine self-healing ability and good mechanical behavior. However, these reported self-healing rubber-based materials are generally faced with difficulties in balancing mechanical properties and self-healing performance, because that the high crosslinking degree favors the mechanical properties while the good self-healing and reprocessable performance is usually achieved for the sulfur-vulcanized carbon-chain rubbers with a low crosslinking degree. To solve the conflict, the carbon-chain rubbers have been vulcanized with only disulfide bond, with disulfide-containing crosslinkers [79-82]. Unfortunately, the functionalized carbon-chain rubbers were used in the proposed approach, such as epoxidized and carboxylated rubbers. It is hardly to be expanded to the common carbon-chain rubbers.
Most recently, Xu's group developed a straightforward strategy to adjust the content of weak sulfide bonds by incorporating vulcanization modifiers into conventional vulcanization formulations [83]. With a high blending contents of vulcanization modifier (benzene-1,4-dithiol), π-π interactions between benzene ring increased entanglement modulus, not only promoting the strain induced crystallization behavior, but also imparting excellent dimensional stability. Moreover, plentiful weak sulfide bonds endowed the self-healing and recyclable property. The representative sample demonstrated a maximum self-healing efficiency of 89.8%, and the remolded samples exhibited a high recovery efficiency of tensile strength, reaching up to 104%, and a recovery of 82.6% for elongation at break. The results indicated that high-performance, self-healing, and recyclable diene-based rubbers could be designed by modulating the composition and content of sulfide bonds in the vulcanization network.
The author declares that he has no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper
Peng Liu: Writing – review & editing, Writing – original draft, Investigation, Funding acquisition, Conceptualization.
This work was financially supported by the Natural Science Foundation of Gansu Province, China (No. 21JR7RA478).
V. Torretta, E.C. Rada, M. Ragazzi, et al., Waste Manag. 45 (2015) 152–160. doi: 10.1016/j.wasman.2015.04.018
A. Mohajerani, H. Kurmus, D. Conti, et al., Sci. Total Environ. 835 (2022) 155269. doi: 10.1016/j.scitotenv.2022.155269
F.N. Archibong, O.M. Sanusi, P. Mederic, N.A. Hocine, Resour. Conserv. Recy. 175 (2021) 105894. doi: 10.1016/j.resconrec.2021.105894
A.A. Basik, J.J. Sanglier, C.T. Yeo, K. Sudesh, Polymers 13 (2021) 1989. doi: 10.3390/polym13121989
L. Bockstal, T. Berchem, Q. Schmetz, A. Richek, J. Clean Prod. 236 (2019) 117574. doi: 10.1016/j.jclepro.2019.07.049
H. Chittella, L.W. Yoon, S. Ramarad, Z.W. Lai, Polym. Degrad. Stab. 194 (2021) 109761. doi: 10.1016/j.polymdegradstab.2021.109761
R. Saputra, R. Walvekar, M. Khalid, N.M. Mubarak, M. Sillanpaa, Chemosphere 265 (2021) 129033. doi: 10.1016/j.chemosphere.2020.129033
P. Wisniewska, S.F. Wang, K. Formela, Waste Manag. 150 (2022) 174–184. doi: 10.1016/j.wasman.2022.07.002
S.Y. Leong, S.Y. Lee, T.Y. Koh, D.T.C. Ang, Waste Manag. 25 (2023) 37–51. doi: 10.1007/s10163-022-01554-y
A. Kumar, R.J. Dhanorkar, S. Mohanty, V.K. Gupta, Mater. Adv. 5 (2024) 7584–7600. doi: 10.1039/d4ma00379a
J.D. Martinez, N. Puy, R. Murillo, et al., Renew. Sust. Energy Rev. 23 (2013) 179–213. doi: 10.1016/j.rser.2013.02.038
T. Zhang, L. Asaro, M. Gratton, N.A. Hocine, J. Environ. Manag. 353 (2024) 120122. doi: 10.1016/j.jenvman.2024.120122
S. Maroufi, M. Mayyas, V. Sahajwalla, Waste Manag. 69 (2017) 110–116. doi: 10.1016/j.wasman.2017.08.020
W. Yang, W.J. Sun, W. Chu, C.F. Jiang, J. Wen, Chin. Chem. Lett. 23 (2012) 363–366. doi: 10.1016/j.cclet.2012.01.006
J.B. Sui, D. Xiang, P. Mou, et al., Adv. Mater. Res. 239-242 (2011) 2503–2510. doi: 10.4028/www.scientific.net/AMR.239-242.2503
P. Soprych, G. Czerski, P. Grzywacz, Energies 17 (2024) 14.
B. Wang, H.W. Ma, K.H. Shen, D. Jun, Y. Li, Chin. Chem. Lett. 23 (2012) 1419–1422. doi: 10.1016/j.cclet.2012.10.018
M.M. Horiks, J. Polym. Sci. 19 (1956) 445–454. doi: 10.1002/pol.1956.120199305
G.K. Jana, R.N. Mahaling, T. Rath, et al., Polimery 52 (2007) 131–136. doi: 10.14314/polimery.2007.131
J.A. Mary, G. Benny, K.N. Madhusoodanan, A. Rosamma, Rubber Sci. 29 (2016) 62–100.
M. Kojima, K. Ogawa, H. Mizoshima, et al., Rubber Chem. Technol. 76 (2003) 957–968. doi: 10.5254/1.3547784
M. Kojima, M. Tosaka, Y. Ikeda, Green Chem. 6 (2004) 84–89. doi: 10.1039/b314137c
M. Kojima, S. Kohjiya, Y. Ikeda, Polymer 46 (2005) 2016–2019. doi: 10.1016/j.polymer.2004.12.053
M. Kojima, M. Tosaka, E. Funami, et al., J. Supercrit. Fluids 35 (2005) 175–181. doi: 10.1016/j.supflu.2005.02.004
Z.J. Liu, X. Li, X.F. Xu, et al., Polym. Degrad. Stab. 119 (2015) 198–207. doi: 10.1016/j.polymdegradstab.2015.05.017
M. Kojima, M. Tosaka, Y. Ikeda, S. Kohjiya, J. Appl. Polym. Sci. 95 (2005) 137–143. doi: 10.1002/app.20806
M. Kojima, Y. Ikeda, Kobunshi Ronbunshu 62 (2005) 242–250. doi: 10.1295/koron.62.242
S.O. Mohaved, A. Ansarifar, S.K. Nezhad, S. Atharyfar, Polym. Degrad. Stab. 111 (2015) 114–123. doi: 10.1016/j.polymdegradstab.2014.11.003
S. Ghorai, S. Bhunia, M. Roy, D. De, Polym. Degrad. Stab. 129 (2016) 34–46. doi: 10.1016/j.polymdegradstab.2016.03.024
S. Rooj, G.C. Basak, P.K. Maji, A.K. Bhowmick, J. Polym. Environ. 19 (2011) 382–390. doi: 10.1007/s10924-011-0293-5
M. Sabzekar, M.P. Chenar, S.M. Mortazavi, et al., Polym. Degrad. Stab. 118 (2015) 88–95. doi: 10.1016/j.polymdegradstab.2015.04.013
J.N. Noel, A.C. Gaumont, J.F. Pilard, I. Dez, ACS Sustainable Chem. Eng. 10 (2022) 159–165. doi: 10.1021/acssuschemeng.1c05228
X. Colom, J. Canavate, K. Formela, A. Shadman, M.R. Saeb, Polym. Degard. Stab. 183 (2021) 109450. doi: 10.1016/j.polymdegradstab.2020.109450
X. Colom, M.R. Saeb, J. Canavate, Express Polym. Lett. 9 (2024) 950–961. doi: 10.3144/expresspolymlett.2024.72
K.A.J. Dukhuis, I. Babu, J.S. Lopulissa, J.W.M. Noordermeer, W.K. Dierkes, Rubber Chem. Technol. 81 (2008) 190–208. doi: 10.5254/1.3548204
P. Sutanto, F.L. Laksmana, F. Picchioni, L.P.B.M. Janssen, Chem. Eng. Sci. 61 (2006) 6442–6453. doi: 10.1016/j.ces.2006.05.024
S. Movahed, A. Ansarifar, G. Zohuri, N. Ghaneie, Y. Kermany, J. Elastom. Plast. 48 (2016) 122–144. doi: 10.1177/0095244314557975
M. Khavarnia, S.O. Movahed, J. Appl. Polym. Sci. 133 (2016) 43363. doi: 10.1002/app.43363
V.V. Rajan, W.K. Dierkes, J.W.A. Noordermeer, R. Joseph, Rubber Chem. Technol. 78 (2005) 855–867. doi: 10.5254/1.3547918
W.C. Wang, K.F. Hao, X.R. Guo, et al., J. Clean Prod. 384 (2023) 135421. doi: 10.1016/j.jclepro.2022.135421
L. Guo, L.C. Bai, J.Y. Zhao, et al., Polymers 16 (2024) 395. doi: 10.3390/polym16030395
M. Karaivanova, A. Kodunova, St. Pavlova, J. Appl. Polym. Sci. 95 (2005) 1002–1013. doi: 10.1002/app.20987
R.R.V.A. Rios, M. Gontijo, V.P. Ferraz, R.M. Lago, M.H. Araujo, J. Braz. Chem. Soc. 17 (2006) 603–608. doi: 10.1590/S0103-50532006000300027
K.A. Dubkov, S.V. Semikolenov, D.P. Ivanov, D.E. Babushkin, V.D. Voronchikhin, Iran Polym. J. 23 (2014) 881–890. doi: 10.1007/s13726-014-0284-1
Y.X. Zhang, Z. Zhang, A.M. Wemyss, et al., ACS Sustainable Chem. Eng. 8 (2020) 9079–9087. doi: 10.1021/acssuschemeng.0c02292
C. Valdes, V. Guzman, C. Ponce, et al., Express Polym. Lett. 19 (2025) 594–609. doi: 10.3144/expresspolymlett.2025.45
O. Buitrago-Sueecun, R. Britto, Iran Polym. J. 29 (2020) 553–567. doi: 10.1007/s13726-020-00818-4
Z. Zhang, J.Y. Li, C.Y. Wan, Y.X. Zhang, S.F. Wang, ACS Sustain. Chem. Eng. 9 (2021) 2378–2387. doi: 10.1021/acssuschemeng.0c08867
Y.L. Xie, A.A. Hassan, P. Song, Z. Zhang, S.F. Wang, Polym. Degrad. Stab. 167 (2019) 292–301. doi: 10.1016/j.polymdegradstab.2019.07.015
D.A. Dubkov, S.V. Semikolenov, D.P. Ivanov, et al., Polym. Degrad. Stab. 97 (2012) 1123–1130. doi: 10.1016/j.polymdegradstab.2012.04.006
S. Gillier-Ritoit, D. Reyx, I. Campistron, A. Laguerre, R.P. Singh, J. Appl. Polym. Sci. 87 (2003) 42–46. doi: 10.1002/app.11661
F. Sadaka, I. Campistron, A. Laguerre, J. -F. Pilard, Polym. Degrad. Stab. 97 (2012) 816–828. doi: 10.1016/j.polymdegradstab.2012.01.019
Z. Luo, X.B. Jiang, W. Yao, Chinese J. Polym. Sci. 34 (2016) 359–366. doi: 10.1007/s10118-016-1746-z
P. Rooshenass, R. Yahya, S.N. Gan, J. Polym. Environ. 26 (2018) 1378–1392. doi: 10.1007/s10924-017-1038-x
T.K.N. Tran, J.F. Pilard, P. Pasetto, J. Appl. Polym. Sci. 132 (2015) 41326. doi: 10.1002/app.41326
P. Liu, C.J. Ai, Ind. Eng. Chem. Res. 57 (2018) 3807–3820. doi: 10.1021/acs.iecr.7b03830
S. Wolf, H. Plenio, Green Chem. 15 (2013) 315–319. doi: 10.1039/C2GC36417D
P.W. Xie, H.X. Zhao, Z.R. Shi, C.O. Pan, P. Liu, J. Clean Prod. 465 (2024) 142826. doi: 10.1016/j.jclepro.2024.142826
F. Sadaka, I. Campistron, A. Laguerre, J.F. Pilard, Polym. Degrad. Stab. 98 (2013) 736–742. doi: 10.1016/j.polymdegradstab.2012.12.018
V. Schallert, C. Slugovc, Macromol. Chem. Phys. 222 (2021) 2100110.
C. Jambou, J.F. Pilard, A.C. Gaumont, I. Dez, Eur. Polym. J. 185 (2023) 111805.
A. Mouawia, A. Nourry, A.C. Gaumont, J.F. Pilard, I. Dez, ACS Sustainable Chem. Eng. 5 (2016) 696–700.
M. Abbas, M. Neubauer, C. Slugovc, Polym. Chem. 9 (2018) 1763–1766. doi: 10.1039/c8py00233a
C. Yang, R.C. Zhang, H.X. Zhao, et al., Polymer 300 (2024) 127022. doi: 10.1016/j.polymer.2024.127022
C.O. Pan, Z.R. Shi, R.C. Zhang, P. Liu, Polym. Degrad. Stab. 225 (2024) 110780.
B. Yougourthen, C. Isabelle, D. Isabelle, Eur. Polym. J. 227 (2025) 113770.
G. Zanchin, G. Leone, Prog. Polym. Sci. 113 (2021) 101342.
P.F. Wu, Q.X. Hu, L.A. Ogunfowora, et al., J. Am. Chem. Soc. 147 (2025) 2960–2977. doi: 10.1021/jacs.4c12730
M. Das, A.R. Parathodika, P. Maji, K. Naskar, Eur. Polym. J. 186 (2023) 111844.
B. Aziz, P. Maji, A.R. Parathodika, K. Naskar, ACS Sustainable Chem. Eng. 12 (2024) 11578–11589. doi: 10.1021/acssuschemeng.4c02047
P. Berto, S. Grelier, F. Peruch, Macromol. Rapid Commun. 38 (2017) 1700475.
M. Das, A.B. Bhattacharya, A.R. Parathodika, K. Naskar, Eur. Polym. J. 174 (2022) 111341.
M.H. Wang, J.H. Zhou, X.L. Jiang, et al., Eur. Polym. J. 146 (2021) 110257.
N.B. Pramanik, G.B. Nando, N.K. Singha, Polymer 69 (2015) 349–356.
H.P. Xiang, H.J. Qian, Z.Y. Lu, M.Z. Rong, M.Q. Zhang, Green Chem. 17 (2015) 4315–4325.
H.P. Xiang, M.Z. Rong, M.Q. Zhang, ACS Sustainable Chem. Eng. 4 (2016) 2715–2724. doi: 10.1021/acssuschemeng.6b00224
A. Kuar, M.M. Fetar, T. Griggs, et al., Commun. Mater. 5 (2024) 212.
J. Cao, S.Q. Li, C.C. Wang, et al., Ind. Eng. Chem. Res. 61 (2022) 13136–13144. doi: 10.1021/acs.iecr.2c02089
L. Imbernon, E.K. Oikonomou, S. Norvez, L. Leibler, Polym. Chem. 6 (2015) 4271–4278.
B. Cheng, X. Lu, J.H. Zhou, R. Qin, Y.L. Yang, ACS Sustainable Chem. Eng. 7 (2019) 4443–4455. doi: 10.1021/acssuschemeng.8b06437
H.P. Xiang, J.F. Yin, G.H. Lin, et al., Chem. Eng. J. 358 (2019) 878–890.
A. Kaur, J.E. Gautrot, G. Cavalli, et al., Polymers 13 (2021) 3347. doi: 10.3390/polym13193347
S.Q. Li, P. Tan, J. Cao, et al., Polymer 332 (2025) 128570.

Scheme 4 Devulcanization of sulfur-vulcanized rubber with BPO by S-S and C-S scission.

Scheme 6 Devulcanization of sulfur-vulcanized rubber with nitric acid/hydrogen peroxide under microwave irradiation.

Scheme 7 Devulcanization of sulfur-vulcanized polybutadiene rubber via ketonization route involving C=C bonds in 1,4-units.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:









 下载:
下载: