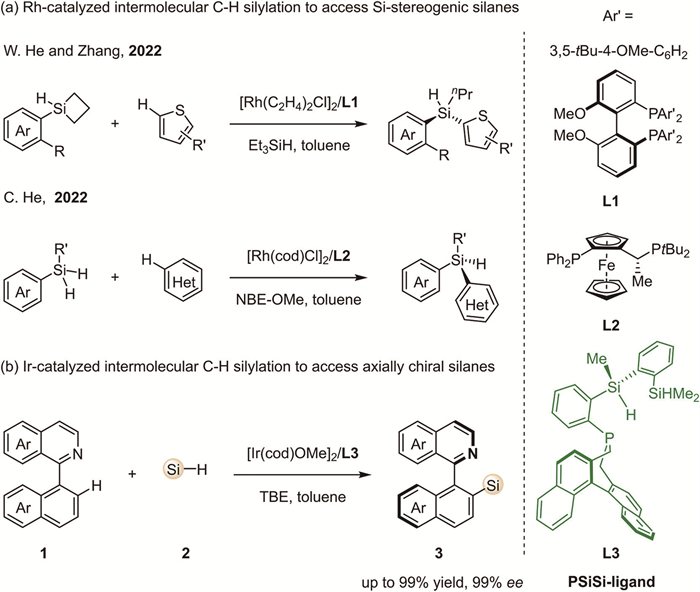
Scheme 1.
Rh- and Ir-catalyzed enantioselective intermolecular C(sp2)-H silylation.

Chiral silicon-containing organic compounds have attracted significant interest due to their unique properties in chemistry, physics, biology, and stereoelectronics, enabling diverse applications in materials science, pharmaceuticals, and agricultural sciences [1-3]. Therefore, the development of efficient methodologies for the synthesis of these compounds is of paramount significance in organic synthesis [4-7]. In this context, transition metal-catalyzed enantioselective C—H silylation reactions have emerged as one of the most atom-economical and straightforward approaches, attracting substantial research interest over the years [8-16]. Nonetheless, while considerable research has been devoted to the intramolecular C—H silylation reactions [17-24], the intermolecular C—H silylation reactions [25-28], particularly in terms of variations in stereoselectivity, remains a relatively unexplored area and presents a significant challenge in this field (Scheme 1). In this context, He, Zhang, and co-workers in 2022 reported the rhodium-catalyzed enantioselective intermolecular C—H silylation of thiophenes with silacyclobutanes (Scheme 1a, up) [25]. In parallel, He and co-workers realized a first example of the rhodium-catalyzed enantioselective intermolecular dehydrogenative C—H silylation of thiophenes and furans (Scheme 1a, below) [26]. Later, Chen, Huang, and co-workers also established a similar strategy for the enantioselective synthesis of acyclic monohydrosilanes via rhodium-catalyzed intermolecular dehydrogenative C—H silylation with heterocyclic compounds [27].
Although rapid progress has been made, these studies have been limited to the asymmetric synthesis of acyclic, silicon-stereogenic silanes (Scheme 1a). Very recently, C. He and co-workers developed the first iridium-catalyzed atroposelective intermolecular C—H silylation of 2-arylisoquinolines, offering a powerful protocol for accessing a series of axially chiral silanes with exceptional yields and enantioselectivities (Scheme 1b) [28]. The key to the success of this transformation lies in the use of the novel chiral PSiSi-ligand. It was found that with a combination of [Ir(OMe)(cod)]2 and chiral PSiSi ligand L3, using the 3,3-dimethylbut-1-ene (TBE) as a hydrogen acceptor, the 2-arylisoquinolines 1 undergo atroposelective C—H silylation with hydrosilanes 2 to afford products 3 in high yields (up to 99%) and excellent enantioselectivities (up to 99% ee). To gain a better understanding of the reaction mechanism, parallel kinetic isotope effect (KIE) experiments were conducted, showing that the Si-H bond cleavage does not occur in the rate-determining step, whereas C—H bond activation is involved in this critical step.
Despite the significant advancement in enantioselective C—H silylation, the mechanistic studies in this area remain scarce and are primarily focused on the intramolecular reactions [29-31]. For instance, we previously reported a computational investigation of iridium-catalyzed intramolecular silylation of unactivated C(sp3)–H bonds, wherein the in situ generated iridium(Ⅲ) silyl dihydride species was found to be the active catalyst [29]. More recently, He and co-workers conducted a combined experimental and computational study on the rhodium-catalyzed intramolecular enantioselective C—H silylation with dihydrosilanes [30]. Following our continuous interest in this field, we therefore decided to investigate the title reaction by means of density functional theory (DFT) calculations [32-42]. Interestingly, the computations unveil a ligand-enabled axial chirality transfer strategy responsible for the observed enantioselectivity, arising from the match/mismatch in axial chirality between the reacting 2-arylisoquinolines and the BINEPINE skeleton of the PSiSi ligand.
All the calculations were performed using Gaussian 09 package [43]. The geometry optimizations were carried out at the B3LYP-D3(BJ) [44-46]. Level of theory with a mixed basis set of SDD for Ir and 6–31G(d) for all other atoms. Frequencies were computed analytically at the same level of theory to confirm whether the structures are minima (no imaginary frequencies) or transition states (only one imaginary frequency). Selected transition-state structures were confirmed to connect the correct reactants and products by intrinsic reaction coordinate (IRC) calculations [47,48]. To obtain better accuracy, energies for the optimized geometries were recalculated with M06 functional [49,50]. using the solution-phase single-point calculations with a larger basis set, which is SDD for Ir and 6–311+G(d,p) for all other atoms. Solvation effects (solvent = toluene, ε=2.3741) were taken into account by performing single-point calculations with the SMD model [51]. The final free energies reported in the article are the large basis set single-point energies corrected by gas-phase Gibbs free energy correction (at 298.15 K). All 3D structures of the optimized geometries were generated using CYLview [52].
The experimentally used 1-(naphthalen-1-yl)isoquinoline 1a and dimethyl(phenyl)silane 2a were selected as the model substrates in the calculations. Experimentally, the stoichiometric reactions suggest that the Ir(Ⅲ) catalyst complex IM1, generated in situ by the reaction of Ir(I) precursor [Ir(cod)OMe]2 with chiral PSiSi-ligand L3 via a dehydrogenative process, is the plausible catalyst species of the reaction [6]. Therefore, the Ir(Ⅲ) catalyst complex IM1 was used as the starting point of the calculations. The sum of the free energies of IM1 and substrates were chosen to be the zero on the relative free energy scale (Fig. 1).
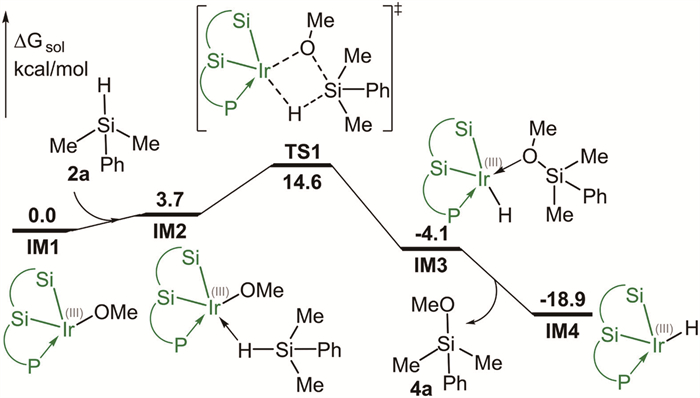
At the outset, the formation of the iridium(Ⅲ) hydride intermediate IM4 was considered (Fig. 1). The process begins with the coordination of 2a to the Ir(Ⅲ) center of IM1 to form intermediate IM2, which was calculated to be endergonic by 3.7 kcal/mol. Then, the σ-bond metathesis was found to occur via transition state TS1 with an energy barrier of 14.6 kcal/mol relative to IM1, giving rise to intermediate IM3 The subsequent dissociation of methoxydimethyl(phenyl)silane 4a from the Ir(Ⅲ) center leads to iridium(Ⅲ) hydride intermediate IM4. The computations show that the formation of intermediate IM4 is highly exergonic by 18.9 kcal/mol. Notably, the intermediate IM4 is identified as the active catalyst species of the reaction (vide infra). It is also worth mentioning that the iridium(Ⅲ) hydride intermediate has previously been proposed as the active catalytic species in the related Ir-catalyzed intramolecular C–H silylation reactions [7].
The calculated energy profile of the most favorable pathway for the intermolecular C—H silylation catalyzed by active catalyst species IM4 is depicted in Fig. 2. Other possible reaction pathways starting from intermediate IM4 were deposited in Fig. S2 (Supporting information). Starting from IM4, the coordination of the C═C double bond of TBE to the Ir(Ⅲ) center delivers π-complex IM5, which was calculated to be exergonic by 9.4 kcal/mol. Then, IM5 undergoes the 2,1-migratory insertion of C═C double bond into the Ir−H bond via transition state TS2, with an energy barrier of 10.6 kcal/mol relative to IM5. The alternative 1,2-migratory insertion was also considered, but it was found to be significantly higher in energy compared to the 2,1-migratory insertion (Fig. S1 in Supporting information for details). The resulting intermediate IM6 is much less stable than IM5 by 9.5 kcal/mol. With the incoming 2a, the ensuing Si-H oxidative addition takes place through transition state TS3 to furnish iridium(Ⅴ) silyl hydride species IM7, requiring an energy barrier of 11.4 kcal/mol relative to IM6 (i.e., 20.9 kcal/mol relative to IM5). The iridium(Ⅴ) silyl hydride species IM7 was found to be relatively unstable, and can readily undergo the C—H reductive elimination via transition state TS4 to form iridium(Ⅲ) silyl species IM8 and release 2,2-dimethylbutane, with an energy barrier of only 3.7 kcal/mol relative to IM7. The computations show that formation of iridium(Ⅲ) silyl species IM8 from iridium(Ⅲ) hydride intermediate IM4 is thermodynamically favored by as much as 24.9 kcal/mol. This substantial energy difference provides the driving force for the overall reaction, underscoring the critical role of TBE in promoting this transformation.
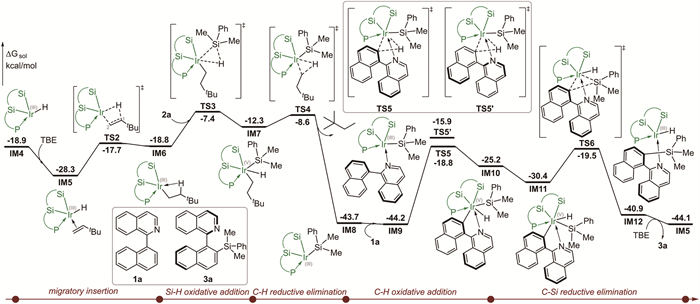
Upon formation of iridium(Ⅲ) silyl species IM8, the next step of the reaction is the C—H oxidative addition, other possible C—H oxidative addition starting from intermediate IM8 (see Fig. S4 in Supporting information for details). The coordination of the incoming 1a to the Ir(Ⅲ) center gives intermediate IM9. The subsequent C—H oxidative addition occurs via transition state TS5, giving rise to iridium(Ⅴ) intermediate IM10. The computations show that this step is endergonic by 19.0 kcal/mol. Finally, the catalytic cycle is completed by the C−Si reductive elimination to form the C(sp2)−H silylation product 3a. However, the C-Si reductive elimination cannot take place directly from IM10, since the H ligand occupies the coordination site between the aryl group and the silyl ligand. Therefore, an isomerization step for the positional change of the hydride ligand is required prior to the C-Si reductive elimination. The computations show that isomerization process is kinetically feasible (Fig. S3 in Supporting information for details). The resulting intermediate IM11, in which the aryl group is adjacent to the silyl ligand, was found to be more stable than IM10 by 5.2 kcal/mol. Then, IM11 can undergo the C-Si reductive elimination via transition state TS6 to afford product-coordinated intermediate IM12, from which the ligand exchange with TBE results in the formation of product 3a and Ir(Ⅲ) complex IM5.
The computations reveal that the C—H oxidative addition constitutes the rate- and enantioselectivity-determining step of the overall reaction, which is in accordance with the KIE experiments. The computed energy difference of 2.9 kcal/mol between TS5 and TS5′ (−18.8 versus −15.9 kcal/mol) corresponds to a predicted ratio of 99:1 at the reaction temperature, which aligns well with the experimentally observed excellent enantioselectivity. The optimized geometries of transition states TS5 and TS5′ are displayed in Fig. 3a. It was found that the key bond distances in two transition states are very different. In particular, the breaking C—H bond in TS5 is greater than that in TS5′ by 0.25 Å (1.72 versus 1.47 Å), implying that transition state TS5 is significantly later than TS5′.
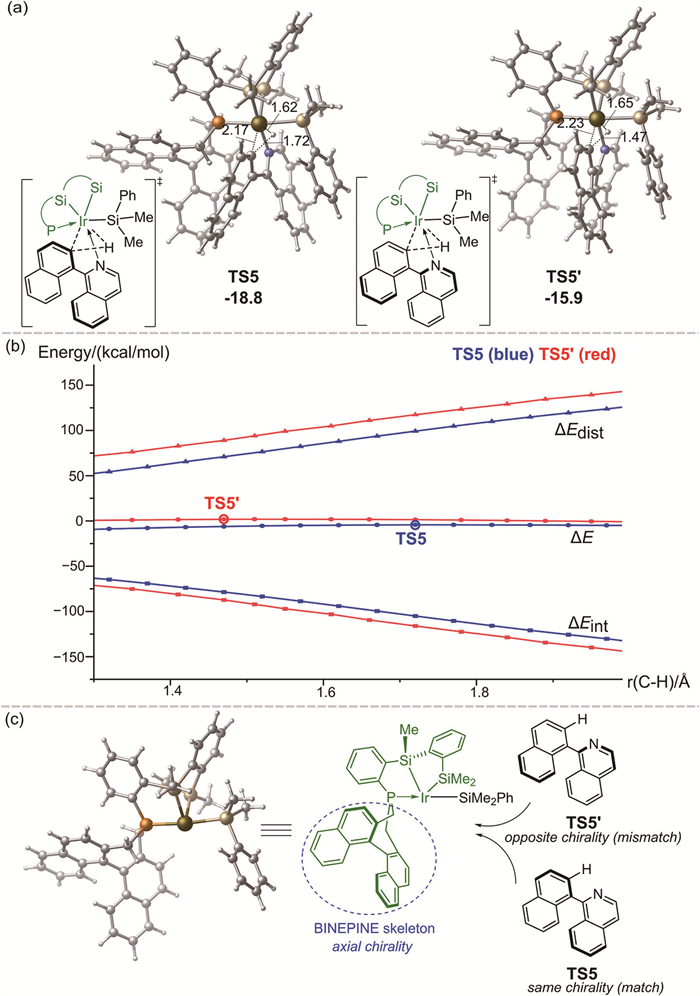
To further elucidate the origins of the enantioselectivity, the distortion/interaction analysis along the reaction pathway was conducted, using the breaking C—H bond distances as a reaction coordinate (Fig. 3b) [53]. The results show that the interaction energies of TS5′are constantly greater than that of TS5 along the reaction pathway, while the distortion energies of TS5′ are higher than that of TS5, being the dominant factor that contributes to the enantioselectivity. Further analysis reveals that the difference in distortion energies primarily arises from 1-(naphthalen-1-yl)isoquinoline 1a moiety (Fig. S5 in Supporting information for details). Thus, the geometric changes of 1a along the reaction pathway play a critical role in determining enantioselectivity. The structural analysis of IM8 moiety shows that the silyl group and the PSiSi ligand on the Ir atom create a compact environment for incoming 1a (Fig. 3c). Importantly, an axially chiral pocket was generated by the BINEPINE skeleton of the PSiSi ligand. In TS5, 1a adopts the same axial chirality as the BINEPINE skeleton, requiring minimal geometric adjustment. By contrast, in TS5′, the axial chirality of 1a is opposite to that of the BINEPINE skeleton, necessitating a larger geometric change to reach the transition state structure. Consequently, the axial chirality of the PSiSi ligand is directly transferred to the reaction, dictated by the match/mismatch in axial chirality between 1a and the BINEPINE skeleton of the PSiSi ligand.
To summarize, we have presented a mechanistic study on the iridium(Ⅲ)-catalyzed intermolecular C—H silylation of 2-arylisoquinolines by means of DFT calculations. The computations show that the iridium(Ⅲ) hydride species is the active catalyst of the reaction. The reaction was found to proceed through the Ir(Ⅲ)/Ir(V) catalytic cycle, consisting of five major steps: (1) Migratory insertion of TBE into the Ir–H bond, (2) Si-H oxidative addition to form iridium(Ⅴ) silyl hydride species, (3) C—H reductive elimination to generate the iridium(Ⅲ) silyl species, (4) C(sp2)-H oxidative addition to yield the iridium(Ⅴ) complex, and (5) C-Si reductive elimination to deliver the final silylation product. The C(sp2)-H oxidative addition constitutes the rate- and enantioselectivity-determining step of the overall reaction. The distortion/interaction and structural analyses reveal a ligand-enabled axial chirality transfer strategy responsible for the observed enantioselectivity. Specifically, the BINEPINE skeleton of the PSiSi ligand creates an axially chiral pocket for the C—H oxidative addition. The match/mismatch in axial chirality between reacting 2-arylisoquinolines and the BINEPINE skeleton of the PSiSi ligand plays a pivotal role in controlling enantioselectivity. The computations should provide important implications for a better understanding of the related C—H silylation reactions and the development of new catalytic systems.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Deng Pan: Writing – review & editing, Writing – original draft, Investigation. Chuan He: Writing – review & editing, Conceptualization. Genping Huang: Writing – review & editing, Writing – original draft, Funding acquisition, Conceptualization.
This work was supported by the National Natural Science Foundation of China (Nos. 22471191, 22073066, and 22271134), and Shenzhen Science and Technology Innovation Commission (Nos. RCJC20221008092723013, JCYJ20230807093104009).
Supplementary material associated with this article can be found, in the online version, at doi:
A.K. Franz, S.O. Wilson, J. Med. Chem. 56 (2013) 388–405. doi: 10.1021/jm3010114
B.A. Kamino, T.P. Bender, Chem. Soc. Rev. 42 (2013) 5119–5130. doi: 10.1039/c3cs35519e
Y. Zuo, Z. Gou, W. Quan, W. Lin, Coord. Chem. Rev. 438 (2021) 213887.
E. Langkopf, D. Schinzer, Chem. Rev. 95 (1995) 1375–1408. doi: 10.1021/cr00037a011
L.W. Xu, L. Li, G.Q. Lai, J.X. Jiang, Chem. Soc. Rev. 40 (2011) 1777–1790. doi: 10.1039/C0CS00037J
C.G. Newton, S.G. Wang, C.C. Oliveira, N. Cramer, Chem. Rev. 117 (2017) 8908–8976. doi: 10.1021/acs.chemrev.6b00692
Y. Wu, P. Wang, Angew. Chem. Int. Ed. 61 (2022) e202205382. doi: 10.1002/anie.202205382
J.F. Hartwig, Acc. Chem. Res. 45 (2012) 864–873. doi: 10.1021/ar200206a
C. Cheng, J.F. Hartwig, Chem. Rev. 115 (2015) 8946–8975. doi: 10.1021/cr5006414
Z. Xu, L.W. Xu, ChemSusChem 8 (2015) 2176–2179. doi: 10.1002/cssc.201500467
S.C. Richter, M. Oestreich, Trends Chem. 2 (2020) 13–27.
B. Li, P.H. Dixneuf, Chem. Soc. Rev. 50 (2021) 5062–5085. doi: 10.1039/d0cs01392g
Y. Ge, X. Huang, J. Ke, C. He, Chem Catal. 2 (2022) 2898–2928.
C.X. Liu, S.Y. Yin, F. Zhao, et al., Chem. Rev. 123 (2023) 10079–10134. doi: 10.1021/acs.chemrev.3c00149
Y. Wu, L. Zheng, Y. Wang, P. Wang, Chem 9 (2023) 3461–3514. doi: 10.1016/j.chempr.2023.09.024
H. Khatua, S. Das, S. Patra, B. Chattopadhyay, Synthesis 55 (2023) 3434–3453. doi: 10.1055/a-2110-4581
L.W. Xu, Angew. Chem. Int. Ed. 51 (2012) 12932–12934. doi: 10.1002/anie.201207932
Q.W. Zhang, K. An, L.C. Liu, et al., Angew. Chem. Int. Ed. 56 (2017) 1125–1129. doi: 10.1002/anie.201609022
D. Mu, W. Yuan, S. Chen, et al., J. Am. Chem. Soc. 142 (2020) 13459–13468. doi: 10.1021/jacs.0c04863
B. Yang, W. Yang, Y. Guo, L. You, C. He, Angew. Chem. Int. Ed. 59 (2020) 22217–22222. doi: 10.1002/anie.202009912
W. Ma, L.C. Liu, K. An, T. He, W. He, Angew. Chem. Int. Ed. 60 (2021) 4245–4251. doi: 10.1002/anie.202013041
S. Chen, D. Mu, P. Mai, et al., Nat. Commun. 12 (2021) 1249. doi: 10.1038/s41467-021-21489-6
Y. Guo, M.M. Liu, X. Zhu, L. Zhu, C. He, Angew. Chem. Int. Ed. 60 (2021) 13887-1389. doi: 10.1002/anie.202103748
Y. Zeng, X.J. Fang, R.H. Tang, et al., Angew. Chem. Int. Ed. 61 (2022) e202214147. doi: 10.1002/anie.202214147
S. Chen, J. Zhu, J. Ke, Y. Li, C. He, Angew. Chem. Int. Ed. 61 (2022) e202117820. doi: 10.1002/anie.202117820
K. An, W. Ma, L.C. Liu, et al., Nat. Commun. 13 (2022) 847. doi: 10.1038/s41467-022-28439-w
D. Mu, S. Pan, X. Wang, et al., Chem. Commun. 58 (2022) 7388–7391. doi: 10.1039/d2cc02307e
B. Yang, J. Gao, X. Tan, Y. Ge, C. He, Angew. Chem. Int. Ed. 62 (2023) e202307812. doi: 10.1002/anie.202307812
M. Zhang, J. Liang, G. Huang, J. Org. Chem. 84 (2019) 2372–2376. doi: 10.1021/acs.joc.9b00117
N. Li, J. Ke, L.Q. Ren, Y. Li, C. He, Chem Catal. 3 (2023) 100799.
C. Karmel, J.F. Hartwig, J. Am. Chem. Soc. 142 (2020) 10494–10505. doi: 10.1021/jacs.0c03301
H. Tamura, H. Yamazaki, H. Sato, S. Sakaki, J. Am. Chem. Soc. 125 (2003) 16114–16126. doi: 10.1021/ja0302937
A.G. Green, P. Liu, C.A. Merlic, K.N. Houk, J. Am. Chem. Soc. 136 (2014) 4575–4583. doi: 10.1021/ja411699u
G. Huang, M. Kalek, R.Z. Liao, F. Himo, Chem. Sci. 6 (2015) 1735–1746. doi: 10.1039/C4SC01592D
J. Jover, F. Maseras, Organometallics 35 (2016) 3221–3226. doi: 10.1021/acs.organomet.6b00562
A. Unnikrishnan, R.B. Sunoj, Chem. Sci. 10 (2019) 3826–3835. doi: 10.1039/c8sc05335a
B.E. Haines, Y. Saito, Y. Segawa, K. Itami, D.G. Musaev, ACS Catal. 6 (2016) 7536–7546. doi: 10.1021/acscatal.6b02317
L. Zhu, X. Qi, Y. Li, et al., Organometallics 36 (2017) 2107–2115. doi: 10.1021/acs.organomet.7b00151
R.L. Zhong, S. Sakaki, J. Am. Chem. Soc. 141 (2019) 9854–9866. doi: 10.1021/jacs.9b01767
R.L. Zhong, S. Sakaki, J. Am. Chem. Soc. 142 (2020) 16732–16747. doi: 10.1021/jacs.0c07239
M. Zhang, H. Wu, Yang J, G. Huang, ACS Catal. 11 (2021) 4833–4847. doi: 10.1021/acscatal.1c00389
L.Y. Bao, J.S. Wang, L. Li, R.L. Zhong. Z.M. Su, J. Org. Chem. 89 (2024) 18047–18059. doi: 10.1021/acs.joc.4c01779
M.J. Frisch, G.W. Trucks, H.B. Schlegel, et al., Gaussian 09, Gaussian, Inc., Wallingford, CT, 2013.
C. Lee, W. Yang, R.G. Parr, Phys. Rev. B. 37 (1988) 785–789. doi: 10.1103/PhysRevB.37.785
A.D. Becke, J. Chem. Phys. 98 (1993) 5648–5652. doi: 10.1063/1.464913
S. Grimme, J. Antony, S. Ehrlich, H. Krieg, J. Chem. Phys. 132 (2010) 154104. doi: 10.1063/1.3382344
K. Fukui, J. Phys. Chem. 74 (1970) 4161–4163. doi: 10.1021/j100717a029
K. Fukui, Acc. Chem. Res. 14 (1981) 363–368. doi: 10.1021/ar00072a001
Y. Zhao, D.G. Truhlar, Acc. Chem. Res. 41 (2008) 157–167. doi: 10.1021/ar700111a
Y. Zhao, D.G. Truhlar, Theor. Chem. Acc. 120 (2008) 215–241. doi: 10.1007/s00214-007-0310-x
A.V. Marenich, C.J. Cramer, D.G. Truhlar, J. Phys. Chem. B 113 (2009) 6378–6396. doi: 10.1021/jp810292n
C.Y. Legault, CYLview, ver. 1.0b, Université de Sherbrooke, 2009
F.M. Bickelhaupt, K.N. Houk, Angew. Chem. Int. Ed. 56 (2017) 10070–10086. doi: 10.1002/anie.201701486

Figure 2 Calculated energy profile of Ir-catalyzed atroposelective intermolecular C-H silylation of 1a.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:


 下载:
下载: